
Artículos originales
Deleciones GJB6-D13S1830 y GJB6-D13S1854 en pacientes con sordera prelingual no sindrómica
GJB6-D13S1830 and GJB6-D13S1854 Deletions in Patients with non-Syndromic Prelingual Deafness

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-05-18 20:34:59
Aprobado: 2024-08-03 16:37:33
Correspondencia: Mercedes Arceo Álvarez. Centro Nacional de Genética Médica. La Habana. fornaris@infomed.sld.cu
RESUMEN
Objetivo: describir la presencia de las deleciones GJB6-D13S1830 y GJB6-D13S1854 en pacientes con sorderas no sindrómicas autosómicas recesivas.
Método: se realizó un estudio descriptivo de corte transversal, a partir 433 pacientes registrados con sorderas no sindrómicas autosómicas recesivas en el Centro Nacional de Genética Médica, en los que se había descartado previamente la presencia de la variante patogénica c.35delG del gen GJB2 en homocigosis. Fue aplicada la técnica de reacción en cadena de la polimerasa multiplex, por el que se describió el genotipo y se determinó su frecuencia.
Resultados: en 13 individuos fueron identificadas deleciones del gen GJB6, 12 en heterocigosis -en siete de ellos la GJB6-D13S1830 y en cinco la GJB6-D13S1854- y uno con la deleción GJB6-D13S1830 en homocigosis. La variante patogénica c.35delG del gen GJB2 había sido previamente identificada en heterocigosis en tres de los pacientes en quienes se identificó la GJB6-D13S1830 y dos en los que se observó la GJB6-D13S1854.
Conclusión: las deleciones GJB6-D13S1830 y GJB6-D13S1854 aparecieron en una baja proporción de los pacientes, con sorderas no sindrómicas autosómicas recesivas, estudiados y fundamentalmente se identificaron en heterocigosis.
Palabras clave: conexina 30; conexina 26; pérdida auditiva, sordera
ABSTRACT
Objective: to describe the presence of GJB6-D13S1830 and GJB6-D13S1854 deletions in patients with autosomal recessive non-syndromic deafness.
Method: a descriptive cross-sectional study was carried out on 433 patients registered with autosomal recessive non-syndromic deafness at the National Center for Medical Genetics, in whom the presence of the pathogenic variant c.35delG of the GJB2 gene had previously been ruled out. homozygosity. The multiplex polymerase chain reaction technique was applied, by which the genotype was described and its frequency was determined.
Results: deletions of the GJB6 gene were identified in 13 individuals, 12 in heterozygosity - in seven of them GJB6-D13S1830 and in five GJB6-D13S1854 - and one with the deletion GJB6-D13S1830 in homozygosity. The pathogenic variant c.35delG of the GJB2 gene had previously been identified in heterozygosity in three of the patients in whom GJB6-D13S1830 was identified and two in whom GJB6-D13S1854 was observed.
Conclusion: GJB6-D13S1830 and GJB6-D13S1854 deletions appeared in a low proportion of the patients with autosomal recessive non-syndromic deafness studied and were mainly identified in heterozygosity.
Key words: connexin 30; connexin 26; hearing loss; deafness
INTRODUCCIóN
La sordera es la discapacidad sensorial más frecuente en el humano, en su forma congénita aparece en cerca de 1 por cada 000 recién nacidos. Se estima que en cerca del 80 % sea de causa genética, la mayoría son sorderas no sindrómicas con herencia autosómica recesiva (SNSAR).(1)
Hasta la fecha se han descrito cerca de 70 genes relacionados con las SNSAR, en los que se han descrito variantes alélicas.(2) A pesar de esta gran heterogeneidad, la mayoría de las SNSAR se deben a mutaciones que yacen en el locus DFNB1, mapeado en 13q11-12. En este lugar se encuentran los genes GJB2 y GJB6 (por sus siglas en inglés); ellos codifican para las conexinas 26 y 30, que forman los canales entre células adyacentes del oído interno, a través de los que ocurren el intercambio de moléculas pequeñas.(3)
La variante patogénica c.35delG, del gen GJB2, es la aparece que en mayor proporción en todas poblaciones como causal de las SNSAR(4) y las deleciones del gen GJB6 -del (GJB6-D13S1830 y del (GJB6-D13S1854)- son las que le siguen en frecuencia en la población española, de donde proviene en parte nuestros ancestros.(5,6,7,8) Se han hallado pacientes con SNSAR en que estas deleciones del gen GJB6, asociadas a variantes patogénicas del gen GJB2, son causa de la pérdida auditiva.(3)
A través de una investigación previa realizada en pacientes cubanos -que clínicamente presentaban una SNSAR- no se concluyó el diagnóstico definitivo en un grupo de ellos, incluidos los heterocigóticos para la mutación c.35delG.(9)
En la medicina actual cada día se acrecienta el interés hacia la prevención, que en las enfermedades hereditarias se logra a través del asesoramiento genético, fundamentado en el diagnóstico preciso. Para las SNSAR las herramientas tradicionales que se han utilizado para el diagnóstico no resultan suficientes, es por ello que la aplicación de pruebas moleculares es imprescindible.
El objetivo de este trabajo fue describir la presencia de las deleciones GJB6-D13S1830 y GJB6-D13S1854 en pacientes con sorderas no sindrómicas autosómicas recesivas (SNSAR).
MéTODOS
Se realizó una investigación descriptiva de corte transversal, a partir de muestras registradas entre los años 2017 y 2023 en el banco ADN del Centro Nacional de Genética Médica. Fueron seleccionadas las muestras correspondientes a los pacientes que presentaban una pérdida auditiva congénita bilateral sensoneural de severa a profunda y tuvieron criterios clínicos para presumir una herencia autosómica recesiva (AR) no sindrómica y que previamente dieron su aprobación en participar en estos estudios.
Fueron excluidos los individuos familiarmente relacionados y los que a través de estudio molecular previo se había concluido su diagnóstico.
De ese modo se estudiaron 433 propósitos (de estos 46 heterocigóticos conocidos previamente para la mutación c.35delG). El ADN se obtuvo, según el protocolo estandarizado en el laboratorio del Centro Nacional de Genética Médica, basado en el método de precipitación salina.(10)
Se utilizó la técnica de reacción en cadena de la polimerasa (PCR) (por sus siglas en inglés) multiplex descrita por del Castillo y cols.(5) Por cada muestra se preparó un volumen total de reacción de 25μl, compuesto por: ADN genómico 100ng, 0,75 U Taq ADN polimerasa, 0,75µl de cada cebador (10pM/μl), 2,5µl dNTP (1mM), 1,5 µl MgCl2 (15mM), Buffer 1X de la enzima: 2,5 µl y H2O: volumen suficiente para completar 25µl por muestra.
Los cebadores utilizados fueron:
Para la amplificación del punto de ruptura del (GJB6-D13S1830):
- GJB6-1R, 5′- TTTAGGGCATGATTGGGGTGATTT-3′
- BKR-1, 5′-CACCATGCGTAGCCTTAACCATTTT-3′
Para la amplificación del punto de ruptura del (GJB6-D13S1854):
- DelBK1, 5′-TCATAGTGAAGAACTCGATGCTGTTT-3′
- DelBK2, 5′-CAGCGGCTACCCTAGTTGTGGT-3′
Para la amplificación del exón 1 del gen GJB6:
- Cx30Ex1A, 5′-CGTCTTTGGGGGTGTTGCTT-3′,
- Cx30Ex1B, CATGAAGAGGGCGTACAAGTTAGAA-3′
La reacción de amplificación consistió en:
- Desnaturalización primaria: 5 minutos a 960C.
Seguido de 30 ciclos de:
- Desnaturalización: 35 segundos a 940C.
- Hibridación: 35 segundos a 560C.
- Elongación: 35 segundos a 720C.
Se concluyó con 7 minutos a 720C, de extensión final.
Esta reacción fue realizada en una máquina de RCP Intelligent heating block IHB 2024, modelo 101.
La separación de los fragmentos de ADN, productos del PCR fueron visualizados a través de la corrida electroforética un gel de agarosa MS al 1,5 %, a 250v por 55 minutos, donde se observó un fragmento de 333pb, correspondiente a la amplificación del exón 1 del gen GJB6. La presencia de la deleción del (GJB6-D13S1830) se identificó cuando se generó un fragmento de 460pb y mientras la deleción del (GJB6-D13S1854) un fragmento de 564pb.
En los que se identificaron las deleciones (GJB6-D13S1830) y del (GJB6-D13S1854), con las que se describió su genotipo, se tomaron en cuenta los resultados previos conocidos respecto a la presencia de la variante c.35delG del gen GJB2 en heterocigosis y su combinación con las deleciones halladas.
Esta investigación se suscribió al proyecto: Detección de dos deleciones en GJB6, del (GJB6-D13S1830) y del (GJB6-D13S1854) en pacientes con sordera no sindrómica, aprobado por el Comité de Ética y el Consejo Científico del Centro Nacional de Genética Médica, a través del dictamen correspondiente. Como parte de una investigación en humanos, se acogió a la a los acápites de la Declaración de Helsinki, atendiendo a su actualización y revisión del 2013.(11)
RESULTADOS
Del total de los 433 individuos no relacionados estudiados, que tenían sordera prelingual no sindrómica, con elementos compatibles a una herencia autosómica recesiva, 420 fueron negativos a las dos deleciones. Se muestra el resultado de la corrida electroforética al explorar dichas deleciones. (Fig. 1).
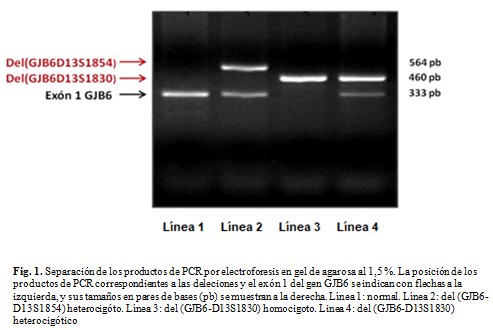
En 13 pacientes se observaron las deleciones estudiadas; uno resultó homocigótico para la mutación del (GJB6-D13S1830), el resto (12) fueron heterocigóticos; entre estos últimos siete presentaron la deleción del (GJB6-D13S1830) y cinco la del (GJB6-D13S1854). De los previamente conocidos como heterocigóticos para la variante patogénica c.35delG del gen GJB2, tres de tuvieron la deleción del (GJB6-D13S1830) y dos la del (GJB6-D13S1854). Los genotipos finales, en los que se tomaron en cuenta los resultados finales de esta investigación, con los previamente conocidos correspondientes a la mutación c.35delG. (Tabla 1).

DISCUSIóN
En el mantenimiento de la estructura y función del oído interno participan diversas proteínas, codificadas por sus correspondientes genes, cada uno de los cuales, pueden presentar diferentes variantes patogénicas causales de pérdidas de la audición neursosensoriales.(12)
Los avances tecnológicos ocurridos en los últimos años han permitido identificar las bases moleculares de las sorderas, comprender su fisiopatología y ampliar las posibilidades en su diagnóstico y manejo. Ello ha sido decisivo fundamentalmente para las SNSAR, en la que su única manifestación es la pérdida de la audición.(2)
El procedimiento aplicado para identificar las variantes patogénicas del gen GJB6 exploradas en esta investigación, es el que ha demostrado mayor utilidad en la práctica.(13,14) Se basa en amplificar los segmentos de ADN que contienen el punto de ruptura de las deleciones del (GJB6-D13s1830) y del (GJB6-D13s1854). Adicionalmente esta técnica permite la identificación de la banda 333 bp, correspondiente su exón 1, que aparece cuando estas variantes patogénicas están ausentes. Ello además de servir para identificar la presencia o no de estas mutaciones, es una forma de validación de los procedimientos moleculares realizados.(5)
El mecanismo por el cual las deleciones del gen GJB6 son causa de pérdida auditiva, cuando están presente en trans con una variante patogénica del gen GJB2, es aún desconocido. El gen GJB6 se localiza adyacente al GJB2 en el locus DFNB1. Se postula que podría ser el resultado de una alteración de la expresión del gen GJB2 localizado en cis a la deleción, a la haploinsificiencia del producto del gen GJB26 o a ambos(13) esto podría explicar los resultados de los pacientes que en esta investigación presentaron variantes patogénicas en heterocigosis compuesta para los genes GJB2 y GJB6.
En la mayoría los individuos en los que se identificaron las deleciones del (GJB6-D13s1830) y del (GJB6-D13s1854) estas se presentaron en heterocigosis. La ocurrencia de las deleciones del gen GJB6 de forma homocigótica han sido encontradas en muy baja frecuencia, también por otros investigadores.(13) Si bien los individuos en que se halló heterocigosis pudieron corresponder a otras variables patogénicas del gen GJB6, lo más frecuente es que se asocie a mutaciones del gen GJB2 no exploradas.(2)
La limitación fundamental de esta investigación consistió en que los estudios para identificar las variantes patogénicas del gen GJB2 solo permitieron el diagnóstico de la mutación c.35delG. Dada la gran heterogeneidad alélica descrita para este gen, donde yacen con más frecuencia las variantes patogénicas causales de pérdidas auditivas, requiere de la aplicación de técnicas de secuenciación que permitan otras posibilidades diagnósticas.(1,2)
Las deleciones GJB6-D13S1830 y GJB6-D13S1854 aparecieron en una baja proporción de los pacientes con SNSAR estudiados y fundamentalmente se identificaron en heterocigosis.
Se agradece a los pacientes por su valiosa contribución con esta investigación.
Conflictos de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Roles de autoría:
1. Conceptualización: Mercedes Arceo Álvarez, Estela Morales Peralta, Teresa Collazo Mesa.
2. Curación de datos: Mercedes Arceo Álvarez, Estela Morales Peralta.
3. Análisis formal: Mercedes Arceo Álvarez, Estela Morales Peralta.
4. Adquisición de fondos: Esta investigación no recibió ningún financiamiento externo.
5. Investigación: Mercedes Arceo Álvarez, Estela Morales Peralta, Yuledmi Perdomo Chacón, Teresa Collazo Mesa.
6. Metodología: Mercedes Arceo Álvarez, Estela Morales Peralta.
7. Administración del proyecto: Mercedes Arceo Álvarez.
8. Recursos: Mercedes Arceo Álvarez, Teresa Collazo Mesa.
9. Software: Estela Morales Peralta.
10. Supervisión: Teresa Collazo Mesa.
11. Validación: Mercedes Arceo Álvarez, Estela Morales Peralta.
12. Visualización: Estela Morales Peralta, Yuledmi Perdomo Chacón.
13. Redacción del borrador original: Estela Morales Peralta.
14. Redacción revisión y edición: Estela Morales Peralta, Yuledmi Perdomo Chacón, Teresa Collazo Mesa.
REFERENCIAS BIBLIOGRÁFICAS
- Adam MP, Fieldman J, Mirzaa MG, Pagon RA, Wallace SE, Bean L, et al. Genetic Hearing Loss Overview. Gene Review [Internet]. Seattle: University of Washington; 2023 [citado 28 Ene 2024]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1434/ [Buscar en Google Scholar]
- Walls D, Azaiez H, Smith R. Hereditary Hearing Loss Homepage [Internet]. Iowa: University of Iowa; 2024 [citado 7 Mar 2024]. Disponible en: https://hereditaryhearingloss.org [Buscar en Google Scholar]
- De Rosa MA, Bernardi MT, Kleppe S, Walz K. Hearing Loss: Genetic Testing, Current Advances and the Situation in Latin America. Genes (Basel). 2024;15(2):178 [Buscar en Google Scholar]
- Hajilari M, Sharifinya A, Khosravi T, Kianmehr A, Taziki MH, Khosravi A, et al. Frequency of c.35delG Mutation in GJB2 gene in Patients with Autosomal Recessive Non-Syndromic Hearing Loss of Five Ethnic Groups in Golestan, Iran. Int J Pediatr. 2023;11(1):17286-98 [Buscar en Google Scholar]
- Del Castillo FJ, Rodríguez M, Álvarez A, Hutchin T, Leonardi E, de Oliveira CA, et al. A novel deletion involving the connexin-30 gene, del (GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J Med Genet. 2005;42(7):588-94 [Buscar en Google Scholar]
- Online Mendelian Inheritance in Man. Gap junction protein, beta-6, GJB6 *604418 [Internet]. Baltimore: Johns Hopkins University; 2022 [citado 6 Mar 2024]. Disponible en: https://omim.org/entry/604418 [Buscar en Google Scholar]
- Online Mendelian Inheritance in Man. Gap junction protein, beta-2, GJB2 *121011 [Internet]. Baltimore : Johns Hopkins University; 2023 [citado 6 Mar 2024]. Disponible en: https://omim.org/entry/121011 [Buscar en Google Scholar]
- Del Castillo I, Moreno MA, Del Castillo FJ, Brownstein Z, Marlin S, Adina Q, et al. Prevalence and evolutionary origins of the del (GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: a multicenter study. Am J Hum Genet. 2003;73(6):1452-8 [Buscar en Google Scholar]
- Morales E, Arceo M, Perdomo Y, Gómez M, Collazo T. Pathogenic variant c.35delG of the GJB2 gene associated with nonsyndromic prelingual deafness. Salud, Cien Tecnol [Internet]. 2024 [citado 11 Jun 2024];4(2):[aprox. 10p]. Disponible en: https://revista.saludcyt.ar/ojs/index.php/sct/article/view/766 [Buscar en Google Scholar]
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16(3):1215 [Buscar en Google Scholar]
- Asociación Médica Mundial. Declaración de Helsinki de la AMM. Principios éticos para las investigaciones médicas en seres humanos. Adoptada por la 64a Asamblea General, Fortaleza, Brasil [Internet]. Brasil: AMM; 2013 [citado 12 May 2024]. Disponible en: https://www.wma.net/es/policies-post/declaracion-de-helsinki-de-la-amm-principios-eticos-para-las-investigaciones-medicas-en-seres-humanos/ [Buscar en Google Scholar]
- Kremer H, Del Castillo I. Genetics of Hearing Impairment. Genes (Basel). 2022;13(5):852 [Buscar en Google Scholar]
- Pandya A, O’Brien A, Kovasala M, Bademci G, Tekin M, Arnos KS. Analyses of del (GJB6-D13S1830) and del (GJB6-D13S1834) deletions in a large cohort with hearing loss: Caveats to interpretation of molecular test results in multiplex families. Mol Genet Genomic Med. 2020;8(4):e1171 [Buscar en Google Scholar]
- Naddafnia H, Noormohammadi Z, Irani S, Salahshoorifar I. Frequency of GJB2 mutations, GJB6-D13S1830 and GJB6-D13S1854 deletions among patients with non-syndromic hearing loss from the central region of Iran. Mol Genet Genomic Med. 2019;7(7):e00780 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129