
Artículos de revisión
Factores genéticos en la carcinogénesis mamaria
Genetic factors for breast carcinogenesis
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2016-09-12 09:43:10
Aprobado: 2016-10-03 15:56:30
Correspondencia: Pedro Enrique Miguel-Soca. Universidad de Ciencias Médicas. Holguín. socahlg@infomed.sld.cu
RESUMEN
Palabras clave: neoplasias de la mama; enfermedades genéticas congénitas; gen BRCA1; gen BRCA2; carcinogénesis
ABSTRACT
Key words: breast neoplasms; genetic diseases inborn; geneBRCA1; gene BRCA2; carcinogenesis
INTRODUCCIÓN
El cáncer de mama es el tumor maligno más frecuente en mujeres, y la causa principal de muerte en todo el mundo con un estimado de 1 700 000 casos y 521 900 fallecidos en 2012, lo que representa el 25 % de los casos de cáncer y 15 % de las muertes por cáncer en las mujeres.1
Se estima que 231 840 féminas serán diagnosticadas de cáncer mamario y más de 40 000 mujeres morirán de esta enfermedad en Estados Unidos, lo que representa la segunda causa de muerte, solo superada por el cáncer de pulmón.2 En 2012, las muertes por este tipo de cáncer representaron 783 000 años de vida potencial perdida y un promedio de 19 años de reducción en la expectativa de vida por cada paciente fallecido.3
Los principales factores de riesgo de este cáncer son: la edad avanzada, los antecedentes familiares de cáncer, la historia personal (mutación positiva BRCA1/BRCA2, biopsia con hiperplasia atípica, biopsia con carcinoma in situ lobular o ductal), la historia reproductiva (menarquia precoz, menopausia tardía, edad tardía del primer embarazo a término o nuliparidad, no lactancia materna o lactar menos de 4 meses), la terapia hormonal de reemplazo con estrógenos y progesterona, el empleo de anticonceptivos orales y los estilos de vida (ganancia de peso, sedentarismo y consumo de alcohol).4-10
La identificación de los factores de riesgo de cáncer de mama permite la aplicación más eficaz de medidas de promoción y prevención de salud, con la consiguiente reducción a largo plazo de la incidencia y mortalidad por este tipo de cáncer. Algunos de estos factores son prevenibles, sobre todo los relacionados con estilos de vida y reproductivos, que es donde se deben ejercer las acciones principales.11-13
En los pacientes con alto riesgo por presentar factores no prevenibles, la búsqueda de nuevos casos en etapas incipientes con métodos de pesquisa activa como la mamografía, sería una medida costo-efectiva favorable; en todos los casos con sospecha de factores genéticos implicados sería importante el diagnóstico precoz mediante tamizaje y el consejo genético a la familia. En el pesquisaje tendría un papel preponderante la atención primaria de salud.14 El conocimiento de estos factores se aplica en ensayos clínicos con fármacos específicos, también es importante para el pronóstico del cáncer mamario.15-19
Los factores de riesgo de cáncer mamario se pueden dividir en dos grupos: factores genéticos y no genéticos. El objetivo de este trabajo es ofrecer una revisión actualizada acerca de los principales genes implicados en la carcinogénesis de mama.
DESARROLLO
Se realizó una amplia revisión de la bibliografía en las principales bases de datos disponibles en la Biblioteca Virtual de Salud de Infomed Nacional (www.sld.cu):
- En SciELO Cuba (http://scieloprueba.sld.cu/scielo.php?script=sci_home&lng=es&nrm=iso) con el descriptor cáncer de mama se recuperaron 36 referencias bibliográficas.
- En SciELO Regional (http://www.scielo.org/php/index.php?lang=es) con los descriptores en inglés: breast cancer and risk factors se encontraron 100 artículos publicados a partir del 2010.
- En Clinical Key (https://www.clinicalkey.es) con los descriptores: breast cancer and risk factors, se encontraron de los últimos 5 años a texto completo 2 863, de los cuales 70 eran revisiones sistemáticas y 267 artículos de revisión.
- En EBSCO (http://search.ebscohost.com) con los mismos descriptores a texto completo con referencias disponibles de los últimos 5 años se encontraron 131 artículos.
- En PubMed (http://www.ncbi.nlm.nih.gov/pubmed) con breast cancer risk se encontraron 6 315 referencias a texto completo de los últimos 5 años referidos a seres humanos (701 revisiones bibliográficas).
El periodo de tiempo de la búsqueda de información y su análisis crítico fue de noviembre del 2015 hasta abril 2016.
Factores de riesgo
El factor de riesgo principal es el sexo porque el cáncer de mama afecta principalmente a las mujeres. En los hombres la incidencia es de alrededor del 1 % de la observada en las mujeres.20 En ellos progresa con más frecuencia hacia un estado avanzado por un diagnóstico tardío, aunque el tratamiento es similar en ambos sexos.
Un segundo factor de riesgo fundamental es la edad, porque la mayoría de los casos de cáncer de mama se diagnostican en mujeres posmenopáusicas de más de 50 años de edad.
El incremento del cáncer de mama en mujeres con activa participación social y profesional implica la necesidad de identificar sus factores de riesgo asociados.21 Del 20-30 % de los nuevos casos se relacionan con factores que inician o modifican el proceso de transformación maligna, entre los que se destacan factores reproductivos, raciales y relacionados con estilos de vida.
Entre los factores de riesgo reproductivos se encuentran la menarquia precoz, la menopausia tardía, la nuliparidad y el primer embarazo a edades tardías, factores que incrementan la exposición estrogénica mamaria. Otros factores no genéticos en la carcinogénesis mamaria son la prescripción a largo plazo de anticonceptivos en mujeres fértiles y la terapia hormonal de reemplazo en mujeres posmenopáusicas.12,22 Los estilos de vida perjudiciales que incrementan este riesgo son la adiposidad, el hábito de fumar y el consumo de alcohol.22
En un número significativo de mujeres con cáncer mamario no se identifican factores de riesgo, lo que sugiere que todavía faltan por determinar factores que contribuyan a la aparición del cáncer.21 Es probable que en estos casos intervengan genes todavía no identificados que incrementan la susceptibilidad a este tipo de neoplasia.
La identificación precisa del papel de los genes en la aparición del cáncer mamario se dificulta por dos razones: primero, transcurre un largo periodo de tiempo entre las alteraciones genéticas y el surgimiento del cáncer; y segundo, la influencia que ejercen los factores relacionados con la reproducción, estilos de vida y agentes ambientales que cambian con el tiempo. Estas consideraciones introducen factores de confusión que deben tenerse en cuenta y explican, al menos, parte de los aspectos controversiales y polémicos del cáncer en general y del mamario en particular. No obstante, con el desarrollo de la biología molecular se han logrado importantes avances en este sentido.
Genes en cáncer mamario
La carcinogénesis comprende alteraciones en el material genético de una célula normal, que trastorna la división celular y la convierte en una célula con una proliferación incontrolable, proceso denominado transformación cancerosa.
La transformación maligna generalmente ocurre en múltiples etapas debido a alteraciones genéticas por mutaciones, aberraciones del número de copias genéticas y la sobreexpresión o silenciamiento de genes, y por alteraciones epigenéticas que provocan cambios en la expresión genética sin modificaciones en la secuencia de bases nitrogenadas del ADN.23 Dos cambios epigenéticos son las modificaciones de histonas y la metilación de promotores de genes. La comprensión de estos mecanismos es relevante en el diagnóstico-pronóstico del cáncer y en el diseño de nuevas estrategias de tratamiento.
Los principales genes implicados en la carcinogénesis mamaria son los oncogenes y los genes supresores tumorales. Los oncogenes son versiones alteradas de genes normales los proto-oncogenes (reguladores positivos de la proliferación celular).24 En su mayoría participan en las vías de transducción de señales y en la supervivencia celular al funcionar como componentes del ciclo celular y de la apoptosis o muerte celular programada. Los supresores del tumor son reguladores negativos de la proliferación celular y, a diferencia de los oncogenes que son dominantes, se comportan como genes recesivos.
La mayoría de los cánceres de mama son positivos a receptores hormonales, diagnosticados después de los 50 años de edad y de etiología multifactorial.25 Las características que sugieren predisposición hereditaria comprenden: aparición en edades tempranas, afectación bilateral, asociación con cáncer ovárico e historia familiar de cáncer de mama u ovario.
Alrededor del 5-10 % de los cánceres mamarios siguen un patrón de herencia autosómico dominante y son hereditarios.26,27 Entre los cánceres de mama hereditarios al menos 30 % se atribuyen a mutaciones germinales en los genes BRCA1 y BRCA2.
La penetrancia de un gen de predisposición al cáncer es su riesgo relativo de causar un tipo particular de cáncer.26 Los genes con alta penetrancia se asocian a un riesgo relativo mayor de 5 y los genes con penetrancia intermedia exhiben un riesgo relativo entre 1,5 y 5. Los alelos de baja penetrancia se presentan generalmente en 10-50 % de la población y confieren menos del 1,5 de incremento de riesgo.27
Los genes de alta penetrancia son: BRCA1, BRCA2, TP53, PTEN, SKT11, CDH1 y MMR y los de penetrancia intermedia son CHEK2, ATM, PALB2 (FANCN), BRIP1 (FANCJ), RAD51C (FANCO), RAD51D, BARD1, MRE11, RAD50, NBS1 y FANCM, en muchos casos responsables en parte del cáncer mamario familiar.26-28
Oncogenes
Los oncogenes se describieron como genes retrovirales que infestaban células normales y las transformaban en células tumorales.29 Sin embargo, la mayoría de los cánceres en seres humanos no tienen un origen viral.
Cada célula tiene dos copias o alelos de cada gen. La activación de los oncogenes a partir de los proto-oncogenes normales tiene acciones dominantes, lo que significa que solo se requiere la activación de un alelo del proto-oncogén de una célula para que se transforme en célula cancerosa. Los mecanismos de activación de oncogenes son diversos: sobreexpresión de un producto genético normal, expresión de una proteína mutante o alteraciones en el reclutamiento o localización de un producto genético normal por interacción con una pareja fijadora mutante o con expresión aberrante.30
Los oncogenes codifican proteínas que controlan la proliferación celular o la apoptosis.31 Estas proteínas se clasifican en 6 categorías: factores de transcripción, proteínas remodeladoras de la estructura de la cromatina, factores de crecimiento, receptores de factores de crecimiento, transductores de señales (proteínas cinasas) y reguladores de la apoptosis o muerte celular programada.
A continuación se describirán los principales oncogenes implicados en el cáncer mamario. (Tabla 1).
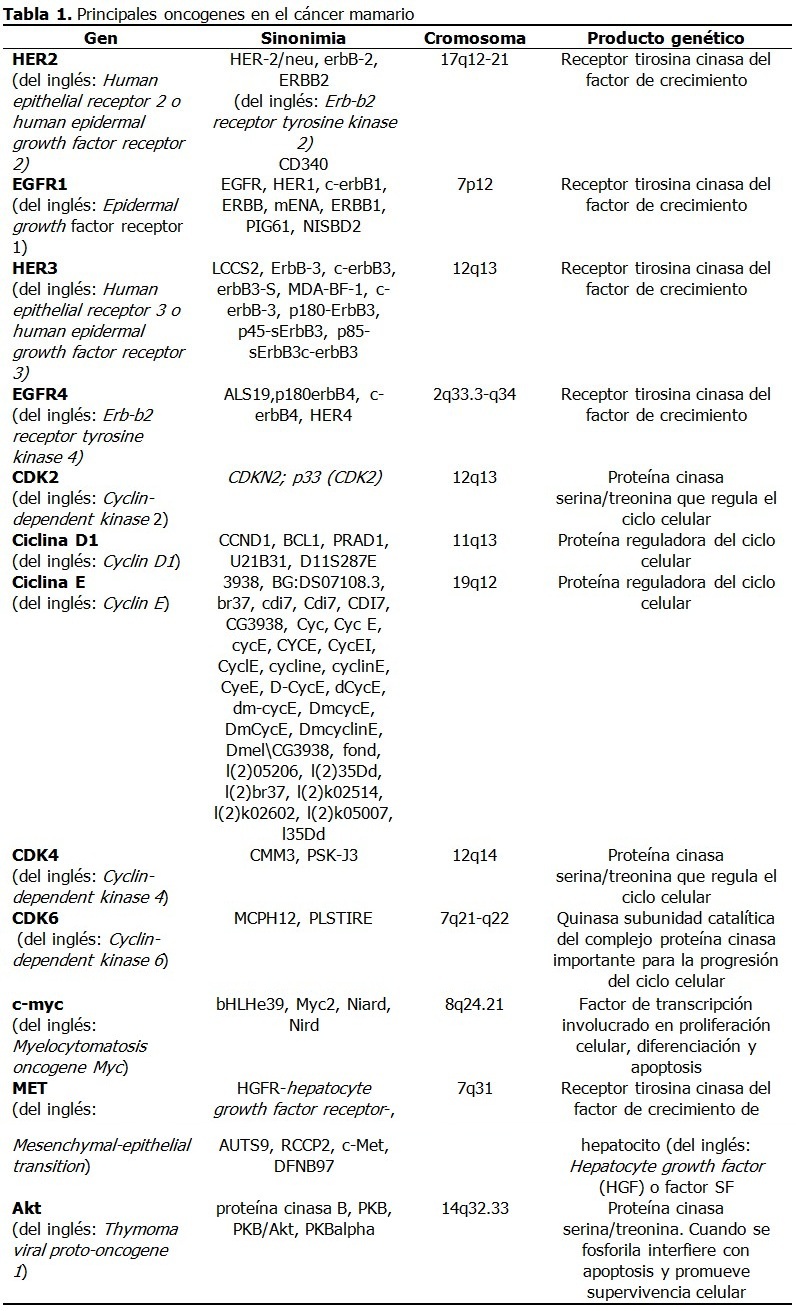
HER2
El gen HER2 codifica un receptor tirosina cinasa de 185-kDa transmembranal del factor de crecimiento.15 La activación del receptor del factor de crecimiento estimula cascadas de transducción de señales que conllevan a la proliferación celular.32 Estas incluyen la quinasa de proteína activada por mitógeno (MAP) y las vías de 3-cinasa (PI3K)/Akt, implicadas en proliferación, angiogénesis, interacciones célula-célula, incremento de motilidad celular, metástasis y resistencia a la apoptosis.15
La sobreexpresión o amplificación de HER-2 se presenta en 15-25 % de los carcinomas mamarios primarios, asociado a una disminución de la supervivencia.31,33 Este descubrimiento ha incrementado el interés por sus aplicaciones diagnósticas y terapéuticas debido a que el 90 % de los cánceres mamarios expresan el receptor de estrógenos α, el receptor de progesterona y el proto-oncogén HER2 y en estos casos el tratamiento con antiestrógenos y agentes diana de HER2 mejora la supervivencia de estos pacientes.34
La expresión de HER2 varía según el subtipo histológico de cáncer mamario con predominio en tumores primarios de origen ductal.15 Este gen se amplifica raramente en lesiones benignas de la mama.
Familia HER
La familia de los receptores del factor de crecimiento epidérmico (EGF) comprende EGFR1, HER2, EGFR3 y EGFR4.35 La sobreexpresión de EGF y sus receptores se produce en cánceres mamarios con mayor incidencia de metástasis, principalmente de EGFR1, HER2 o EGFR3.28,36 Los pacientes con sobreexpresión de EGFR4 tienen mejor evolución.28
Las mutaciones de EGFR4 se asocian a tumores malignos. Aunque aislado de células transformadas de mama, su expresión no está bien demostrada en este tipo de neoplasia.37
EGFR1, expresado frecuentemente en el cáncer de pulmón, cabeza y cuello y en menor medida en el cáncer mamario, codifica una glicoproteína transmembranal que al unirse al factor de crecimiento epidérmico (EGF) (por sus siglas en inglés) induce la dimerización del receptor y la autofosforilación de tirosina, lo que conlleva a la proliferación celular.
CDK2
El ciclo celular se regula principalmente por cinasas dependientes de la ciclina (del inglés: Cyclin-dependent kinases -CDKs), un grupo de proteínas controladas positivamente por ciclinas y negativamente por inhibidores de quinasas dependientes de la ciclina (del inglés: Cyclin-dependent kinase inhibitors -CKIs).38 Cuando las CDKs se regulan en alta, las células proliferan incontrolablemente y son resistentes a la apoptosis.
Las ciclinas se comportan como oncogenes y estimulan el crecimiento celular. Cuando se activan, las CDKs promueven la fosforilación de otras proteínas, especialmente del retinoblastoma (pRb), que permite que la célula pase del estado de reposo, G0, al estado activo y la mitosis.
CDK2 es importante en la transformación maligna de las células epiteliales mamarias, debido probablemente a su unión con ciclina D1 o ciclina E.38 La inhibición de la actividad de CDK2 podría restringir la proliferación de las células cancerosas, incluyendo aquellas resistentes a la terapia endocrina.
Ciclina D1
Este gen se sobreexpresa en 40 % - 50 % de los cánceres mamarios invasivos y se amplifica en 10 % - 20 % de todos los casos.15 Cuando la ciclina D1 se une a su pareja de CDK, se fosforila pRb, se libera el factor transcripcional E2F y se inducen la proteínas necesarias para la síntesis de ADN. Esta ciclina forma un complejo y funciona como subunidad reguladora de CDK4 o CDK6, cuya actividad se requiere para la transición G1/S.
El control aberrante del ciclo celular es el sello distintivo del cáncer. La activación por mitógenos de la síntesis de ciclina D1 estimula la expresión aberrante de factores de crecimiento o sus receptores, lo que activa a las células a producir ciclina D1 de forma autocrina.39,40 Estas vías también se activan constitutivamente debido a la sobreexpresión de moléculas de señalización que participan en la expresión de ciclina D1 como proteínas Ras, que frecuentemente se sobreexpresan en cáncer mamario y se asocian a peor pronóstico.
Ciclina E
El gen que codifica a la ciclina E se amplifica raramente en cáncer mamario (~2 %); aunque, la sobreexpresión y alteraciones de las vías de degradación acumulan isoformas de bajo peso molecular, presentes en 20 % - 30 % de los cánceres de mama.15 De forma similar a la ciclina D1, la sobreexpresión de ciclina E provoca la hiperfosforilación de pRb y el incremento de la actividad proliferativa celular.
CDK4
Un reciente estudio encontró que las mutaciones de CCND1 y CDK4 incrementaban el riesgo de cáncer de mama y podrían servir como biomarcadores para el diagnóstico precoz de este cáncer.39,41
CDK6
CDK6 codifica una quinasa que actúa como subunidad catalítica del complejo proteína cinasa y es importante para la progresión del ciclo celular. La actividad de esta quinasa aparece primero a mediados de la fase G1 y se controla por las subunidades reguladoras ciclinas D y miembros de la familia INK4 de inhibidores de CDK. Esta quinasa como complejo CDK4 fosforila y regula la actividad de Rb y su expresión está regulada en alta en algunos tipos de cánceres.42
C-myc
El oncogén c-myc codifica una fosfoproteína nuclear multifuncional de 65 kDa y 439 aminoácidos, que actúa como un factor de transcripción que regula la transcripción de genes específicos y está involucrado en la proliferación celular, la diferenciación y la apoptosis. La proteína c-Myc se amplifica y sobreexpresa en 15 % - 25 % de los tumores mamarios y en algunos casos, se asocia a peor pronóstico y rasgos clínicos más agresivos.15,43,44
MET
El oncogén MET codifica un receptor tirosina cinasa del factor de crecimiento de hepatocito (del inglés: Hepatocyte growth factor-HGF-), o factor SF.45,46 El ligando unido a MET induce la dimerización y autofosforilación citoplasmática del dominio cinasa del receptor, lo que favorece una cascada de señalización intracelular involucrada en la invasión celular. En el tejido normal, las vías reguladas por MET tienen un papel clave en la embriogénesis, angiogénesis y crecimiento celular.
La activación del eje HGF/MET es un proceso relevante para el inicio y progresión del cáncer, lo que favorece la diseminación de las células cancerosas. MET con frecuencia está desregulado en el cáncer por diferentes mecanismos como amplificación genética o incremento del número de copias, mutaciones somáticas o germinales o sobreexpresión del receptor.45 Estos eventos se han descrito en un amplio espectro de tumores con comportamiento más agresivo. Por ejemplo, MET se sobreexpresa en 20 -30 % de los cánceres de mama y se correlaciona con peor pronóstico.45,47,48
Akt
Este gen codifica un miembro de la familia genética Akt serina treonina proteína cinasa que también incluye Akt2 y Akt3.49 La proteína cinasa B una vez activada por fosforilación juega un papel importante en múltiples procesos celulares. En particular Akt fosforilada (pAkt) interfiere con las funciones apoptósicas de la célula y promueve la supervivencia y migración de las células, lo que favorece la carcinogénesis.49 Esta cinasa es un efector importante de la vía de quinasa de fosfatidilinositol 3 (del inglés: Phosphatidylinositol 3-kinase --PI3K), que media los efectos de factores de crecimiento como factor de crecimiento derivado de plaquetas (del inglés: Platelet-derived growth factor-PDGF), factor de crecimiento epidérmico (Epidermal growth factor) (EGF), Insulin and insulin-like growth factor I (IGF-I).
PI3Ks generan principalmente fosfolípidos que sirven de transductores de señales originados por receptores de tirosina cinasas y receptores acoplados a proteína G.50 La sobreexpresión de Akt se observa con frecuencia en tumores malignos. En el cáncer mamario se asocia la sobreexpresión de este gen con peor pronóstico, aunque existen resultados contradictorios.49
Genes supresores tumorales
Los genes supresores tumorales son genes cuya pérdida de función promueven la malignización, por constituir habitualmente reguladores negativos del crecimiento y otras funciones celulares que afectan el potencial invasivo y metastásico como, la adhesión celular y el control de la actividad de proteasas.
Aunque las anormalidades hereditarias representan una minoría de los casos de cáncer mamario, aparecen mutaciones en la línea germinal (presentes en todas las células) en los genes supresores tumorales, aunque pueden aparecer mutaciones somáticas (presentes solo en células malignas) esporádicas. En ambos casos el tumor contiene una mutación en un alelo y una delección o pérdida del alelo remanente origina el fenotipo maligno. A veces, no ocurren mutaciones en estos genes sino otros mecanismos que interfieren con su función y expresión como la metilación del promotor que suprime su transcripción e incrementa su degradación o alteraciones en otras proteínas que interactúan con el producto genético.15
Para que se exprese el fenotipo canceroso deben alterarse las dos copias o alelos de los genes supresores tumorales por su carácter recesivo.24
La mayor información sobre los genes supresores tumorales proviene de los síndromes cancerosos hereditarios. (Tabla 2).
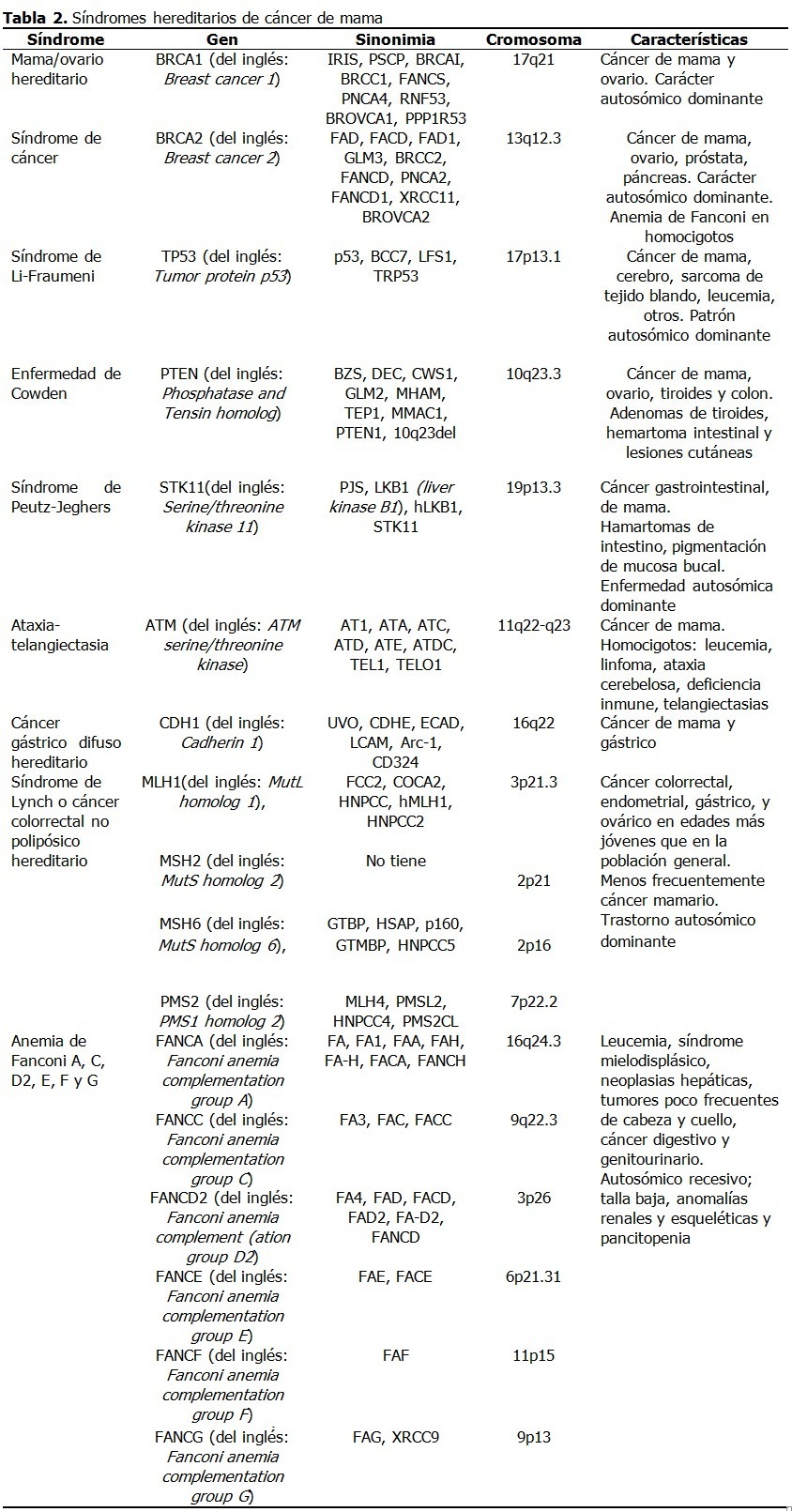
La genética del cáncer mamario se volvió un tema muy publicitado en la prensa de Estados Unidos cuando en mayo de 2013, la actriz Angelina Jolie publicó su decisión de hacerse mastectomía bilateral al conocer su condición de portadora de una mutación en BRCA.51
BRCA1
Las mutaciones en los genes BRCA1 y BRCA2 representan la mayoría de los cánceres de mama heredados con carácter autosómico dominante. Estos genes actúan como genes supresores tumorales, cuyos productos participan en el mantenimiento de la integridad del ADN y en regulación de la transcripción.
Este gen se identificó en 1990 y se conoció como BRCA1 en 1994.52 Se estima que 0,12 % de la población general es portadora de mutaciones de BRCA1, aunque esta tasa es más alta en ciertos grupos como en los judíos Ashkenazi.15
Las mutaciones de BRCA1 aparecen en alrededor del 5 % de los casos de cáncer mamario en mujeres menores de 40 años, pero se eleva a más del 90 % en pacientes con antecedentes patológicos familiares de cáncer de mama u ovario.15 El riesgo de por vida para este cáncer en pacientes con mutaciones de BRCA1 se estima en 49 - 73 % a los 50 años, y en 71 - 87 % a los 70, con un 20 - 30 % de riesgo de cáncer ovárico.15
BRCA-1 codifica una proteína de 1 863 aminoácidos que interactúa con la ARN polimerasa II y con los complejos desacetilasa de histonas.28 BRCA1 juega un papel en la transcripción, reparación del ADN y en la ubiquitinación (mecanismo de degradación de proteínas). La proteína BRCA1 también se combina con otras proteínas para detectar el daño del ADN para formar un complejo proteico oligomérico conocido como BASC (del inglés: BRCA1-associated genome surveillance complex), cuyos componentes pueden mutarse en algunos cánceres.28
BRCA2
El gen BRCA2 muestra rasgos similares a BRCA1, aunque difiere en estructura. La proteína BRCA2 se une a Rad51, lo que garantiza la alta fidelidad de la reparación del ADN. Ambos genes mutados confieren un alto riesgo de cánceres de mama y ovario.15
Aunque el 20-30 % de las mujeres con cáncer de mama tienen al menos un familiar con historia de cáncer mamario, solo el 5-10 % de estas mujeres tienen una predisposición hereditaria identificable. Mutaciones de estos genes son responsables del 3-8 % de todos los casos de cáncer mamario y de 15-20 % de los casos familiares.
Las mujeres con mutaciones hereditarias de BRCA1 y BRCA2 tienen un riesgo de por vida de 50-80 % de cáncer de mama. Las mutaciones de BRCA1 se ven en 7 % de familias con múltiples cánceres de mama y en 40 % de familias con cáncer de ovario y de mama. Las mutaciones de BRCA1 tienen un riesgo de 40 % de cáncer ovárico. Las portadoras de estas mutaciones con cáncer tienen una mayor probabilidad de presentar tumores mamarios triple negativo o subtipo basal, además de cáncer colónico y prostático.
Las mutaciones de BRCA2 se identifican en 10-20 % de familias con alto riesgo de cánceres de mama y ovario y en 2,7 % de mujeres con cáncer precoz de mama. El riesgo de estas mujeres es 10 % de desarrollar cáncer de ovario. El BRCA2 también es un factor de riesgo de cáncer mamario en hombres, aunque estas mutaciones se asocian con otros tumores malignos.
El síndrome hereditario de cáncer mamario y ovárico se vincula a mutaciones de BRCA1 y BRCA2.53 Las que tienen mutaciones de BRCA1 tienen mayor riesgo de cáncer ovárico y son más jóvenes. También los pacientes con mutaciones de BRCA1 tienen más riesgo de presentar otros tumores como: cervical, uterino, pancreático, gástrico y de próstata, mientras que los pacientes con mutaciones de BRCA2 tienen mayor riesgo de melanomas, tumores de vesícula, conducto biliar, próstata y estómago.53 Estas pacientes son candidatas para estrategias de reducción de riesgo como la ooforectomía y mastectomía y cuando se diagnostican con cáncer mamario, tienen mayor riesgo de presentar cáncer en la mama contralateral, lo que tiene importancia en el seguimiento médico.51,54
PT53
El gen codifica una proteína cuya principal función es actuar como factor de transcripción.53,55 TP53 es un gen supresor tumoral involucrado en la detención del crecimiento o división celular, la apoptosis y la reparación del ADN. La pérdida de función de PT53 como guardián del genoma desregula estos procesos.
Las mutaciones de PT53 aparecen en la mitad de los cánceres en seres humanos y en alrededor del 20-30 % de los cánceres primarios de mama.15 PT53 juega un importante rol en el pronóstico del cáncer mamario pues su sobreexpresión provoca una mala respuesta a la quimioterapia, por constituir tumores invasivos, poco diferenciados y con alto grado de malignidad.34
El síndrome de Li-Fraumeni, una rara predisposición familiar que aparece en la segunda o tercera décadas de la vida, se debe a múltiples mutaciones de la línea germinal de PT53.53,55 El riesgo de cáncer de estos pacientes se estima en 50 % para la cuarta década de vida y de 90 % para la octava.15 Este síndrome es responsable de alrededor del 1% de los casos de cáncer mamario familiar, se observa cáncer bilateral en hasta el 25 % de los pacientes y la susceptibilidad al cáncer se transmite con un patrón autosómico dominante.26,52
PTEN
Este gen supresor tumoral codifica una fosfatidilinositol-3,4,5-trifosfatasa que contiene un dominio semejante a tensina y un dominio catalítico similar al que le da especificidad dual a las proteína tirosina fosfatasas. A diferencia de la mayoría de tirosina fosfatasas, esta proteína desfosforila preferentemente sustratos de fosfoinositidos. El gen codifica una fosfatasa que sirve como un regulador negativo de Akt.15
PTEN es un importante interruptor de la carcinogénesis debido a su actividad fosfatasa de fosfatidilinositol-3-cinasa (PI3K).53 No se conoce bien la función de PTEN, aunque su disfunción lo incapacita para parar el ciclo celular y la apoptosis, lo que origina una supervivencia celular anormal.
La enfermedad de Cowden o síndrome de hamartoma tumoral de PTEN es un raro síndrome genético causado por mutaciones germinales en PTEN que se relaciona con hemartoma intestinal, lesiones cutáneas y cáncer de tiroides.53 Las mujeres con esta enfermedad tienen 30 % de prevalencia de cáncer mamario y presentan con más frecuencia lesiones benignas de mama como fibroadenomas y lesiones fibroquísticas.
Las lesiones mucocutáneas como las queratosis acrales y pápulas papilomatosas son rasgos patognomónicos de este síndrome y alrededor del 80 % de las personas afectadas presentarán mutaciones de PTEN y entre quienes no tienen mutaciones detectables, la mitad tendrán mutaciones de su promotor.53
ATM
ATM es un gen supresor tumoral cuyo producto, una cinasa de treonina/serina, tiene un rol central en el mantenimiento de la integridad del genoma por activación de los puntos de control del ciclo celular y en la reparación del ADN.56 ATM también tiene un papel en la detención del ciclo celular, apoptosis y senescencia, lo que evita la inestabilidad genómica. ATM es un gen de susceptibilidad al cáncer de mama con baja penetrancia y baja frecuencia.56
La pérdida de ambos alelos de ATM provoca ataxia telangiectasia, un síndrome de progresiva degeneración cerebelar con ataxia, fragilidad de los vasos sanguíneos, defectos inmunológicos, y predisposición a tumores como el cáncer mamario.15
Por otro lado, los portadores heterocigóticos de mutaciones tienen dos veces mayor riesgo de cáncer de mama comparados con la población general.53 En mujeres <50 años, el riesgo se incrementa 5 veces. La disminución de la expresión de la proteína ATM se asocia con rasgos más agresivos en casos esporádicos de cáncer mamario.
SKT-11
El gen SKT11 codifica la proteína quinasa serina/treonina 11, se identifica como un gen supresor tumoral en muchos cánceres como el de mama y su baja expresión se relaciona con un peor pronóstico. 53,57
Las mutaciones germinales de SKT11 causan el síndrome de Peutz–Jeghers, una rara enfermedad autosómica dominante, caracterizada por múltiples pólipos hemartomatosos y pigmentaciones mucocutáneas de los labios, mucosa bucal y dedos, cuyas lesiones desaparecen durante la pubertad, excepto en la mucosa oral.53 Los pólipos del tracto gastrointestinal pueden provocar obstrucción intestinal, la mayoría se localiza en intestino delgado.
Estas personas tienen un mayor riesgo de cáncer colorrectal, mama, intestino delgado, páncreas, estómago y ovario. Las mutaciones de SKT11 se detectan en alrededor de 70–80 % de los pacientes con el síndrome.53 En estas pacientes se estimó un riesgo relativo 15 veces más alto de cáncer mamario comparado con la población general.52
CDH1
CDH1 es gen supresor tumoral que con frecuencia en el cáncer mamario metastásico es silenciado, mutado o desregulado.58 Este gen codifica una molécula de adhesión epitelial, dependiente de calcio, que juega un papel importante en el establecimiento y mantenimiento de conexiones intercelulares. En comparación con el epitelio normal, las células cancerosas muestran un decrecimiento de la adhesión intercelular mediada por caderina.59
Las mutaciones germinales de CDH1 se asocian al cáncer gástrico difuso hereditario (del inglés: Hereditary diffuse gastric cancer -HDGC). Un 30 % de las familias con mutaciones de CDH1 presentan mujeres con cáncer mamario lobular. El riesgo acumulativo de este tipo de cáncer en portadoras de mutaciones en familias con este cáncer hereditario alcanza el 39 % a los 80 años.53
MMR
Los genes MLH1, MSH2, MSH6 y PMS2 codifican proteínas componentes del sistema de reparación por mal apareamiento del ADN, MMR (del inglés: DNA mismatch repair) que reparan los errores ocurridos durante la replicación.53 Las mutaciones germinales en estos genes causan el cáncer colorrectal no polipósico hereditario o síndrome de Lynch (del inglés: Hereditary Non Polyposis Colorectal Cancer), un trastorno autosómico dominante que se caracteriza por un sustancial riesgo de cánceres como colorrectal, endometrial, gástrico, y ovárico en edades más jóvenes que en la población general. Se debate si el cáncer mamario es parte del síndrome de Lynch.53
La genética en el diagnóstico, pronóstico y tratamiento del cáncer
El fenotipo de cáncer mamario asociado a una mutación BRCA1 es distinto del correspondiente al cáncer mamario esporádico, mientras que el cáncer de mama asociado a una mutación BRCA2 tiene un fenotipo más parecido al de los tumores esporádicos. También se investiga el perfil genético de los polimorfismos de nucleótido único para identificar la predisposición genética al cáncer. Los estudios de asociación en todo el genoma ofrecen información útil respecto al riesgo y permiten desarrollar tratamientos personalizados frente al cáncer.
La clasificación histológica de los carcinomas de mama no refleja la heterogeneidad biológica tumoral, ni permite identificar los pacientes que presentarán mejores respuestas y beneficios con las diferentes terapéuticas.60 La diversidad clínica y pronóstica de los carcinomas de mama que son semejantes y homogéneos en cuanto a sus factores pronósticos clásicos, se establece a nivel molecular, al expresar distintos genes que les confieren variabilidad biológica y pronóstica. Durante los últimos años, el estudio de estos genes ha permitido comprender el comportamiento biológico del cáncer de mama e individualizar el pronóstico y el tratamiento de algunos pacientes.
Con el análisis genómico se clasifican los carcinomas de mama en cinco subtipos: luminal A y B, HER2-positivo, basal y similar a la mama normal. Los carcinomas de tipo luminal tienen mejor pronóstico y se caracterizan por expresar el gen del receptor de estrógenos, por lo que estos tumores pueden tratarse con tamoxifeno o inhibidores de la aromatasa, pero muestran una baja respuesta a la quimioterapia.
El carcinoma de mama HER2-positivo muestra expresión aumentada de genes asociados a c-erbB-2 y suele asociarse a otros marcadores de mal pronóstico. Aunque muestran una mejor respuesta a la quimioterapia y cerca de 50 % responde al tratamiento con trastuzumab, el pronóstico es malo. Otro inhibidor del receptor HER2 como lapatinib puede ser útil en estos pacientes.61
El subtipo basal se caracteriza por la sobreexpresión de citoqueratinas características de la capa basal y de genes de la proliferación celular. Estos tumores presentan mutaciones en TP53, sobreexpresan el receptor del factor de crecimiento epidérmico y se caracterizan por la ausencia de expresión de receptor de estrógenos y de HER2. Este subtipo se asocia a la mutación BRCA1 y presenta el comportamiento más agresivo a pesar de su alta sensibilidad a la quimioterapia.
Aunque el perfil de expresión génica constituye la mejor forma de clasificar los carcinomas de mama, en la mayoría de los hospitales y áreas de salud su uso se encuentra limitado porque constituyen técnicas caras y difíciles de aplicar. En la práctica, la mayor parte de los diagnósticos se realizan mediante otras técnicas como la inmunohistoquímica que, con un limitado número de marcadores (receptor de estrógenos, c-erbB-2, etc.), pueden clasificar los carcinomas mamarios en subtipos equivalentes a aquellos basados en perfiles de expresión génica. La ventaja de estos estudios es que utiliza marcadores disponibles en la mayoría de los Servicios de Anatomía Patológica y puede aplicarse sobre material archivado.
El 70 % de los tumores de mama son positivos a receptores de estrógenos.62 En estos casos, la terapia endocrina es el más efectivo tratamiento, lo que se alcanza con el bloqueo de la fijación de ligandos a los receptores (tamoxifeno y otros moduladores selectivos), regulando en baja los receptores (fulvestrant) o con el bloqueo de la síntesis de estrógenos con inhibidores de aromatasa y agonistas de la hormona liberadora de LH.56,62 En el caso de resistencia tumoral a la terapia endocrina, el conocimiento de la genética molecular del cáncer es de vital importancia para entender los mecanismos implicados y diseñar terapias más adecuadas.
En las mujeres posmenopáusicas, la fuente principal de estrógenos son los andrógenos suprarrenales por acción de la aromatasa en los tejidos periféricos como mamarios o adiposos. Los inhibidores de la aromatasa (anastrozol, letrozol y exemestrano) inhiben esta conversión, lo que disminuye la síntesis de estrógenos en estas mujeres. Los ensayos clínicos demuestran que la eficacia de los inhibidores de la aromatasa es similar o superior a la del tamoxifeno, con aceptables efectos adversos.
CONCLUSIONES
El cáncer de mama es una enfermedad genética con elevada prevalencia y mortalidad, donde convergen factores de riesgo genéticos, y relacionados con la reproducción, ambiente y estilos de vida, en una compleja red de interrelaciones.
Los dos mecanismos básicos de la carcinogénesis mamaria son: la activación de los oncogenes dominantes y la inactivación de los genes supresores tumorales recesivos debido a alteraciones genéticas y epigenéticos.
El tema es controversial y con muchos aspectos polémicos que requerirán estudios posteriores para esclarecerlos, pero de vital importancia para establecer mejores opciones de prevención, diagnóstico, pronóstico y tratamiento del cáncer mamario.
REFERENCIAS BIBLIOGRÁFICAS
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin [revista en Internet]. 2015 [citado 22 Sep 2016];65(2):[aprox. 20p]. Disponible en: http://onlinelibrary.wiley.com/doi/10.3322/caac.21262/full [Buscar en Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin [revista en Internet]. 2015 [citado 25 Sep 2016];65(1):[aprox. 15p]. Disponible en: http://onlinelibrary.wiley.com/doi/10.3322/caac.21254/full [Buscar en Google Scholar]
- Oeffinger KC, Fontham ETH, Etzioni R, Herzig A, Michaelson JS, Shih YCT, et al. Breast Cancer Screening for Women at Average Risk: 2015 Guideline Update From the American Cancer Society. JAMA [revista en Internet]. 2015 [citado 4 Sep 2016];314(15):[aprox. 15p]. Disponible en: http://jama.jamanetwork.com/article.aspx?articleid=2463262#tab6 [Buscar en Google Scholar]
- Coronado GD, Beasley J, Livaudais J. Alcohol consumption and the risk of breast cancer. Salud Pública Méx [revista en Internet]. 2011 [citado 15 Sep 2016];53(5):[aprox. 8p]. Disponible en: http://www.scielo.org.mx/scielo.php?script=sci_arttext&pid=S0036-36342011000500012&lng=es [Buscar en Google Scholar]
- Cuenca C, Despaigne AE, Beltrán Y. Factores de riesgo de cáncer de mama en mujeres pertenecientes a un consultorio médico del Centro Urbano ¨José Martí¨. MEDISAN [revista en Internet]. 2013 [citado 5 Ene 2016];17(9):[aprox. 6p]. Disponible en: http://scieloprueba.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192013000900005&lng=es [Buscar en Google Scholar]
- Núñez AC, Frómeta CI, Rubio T. Factores ambientales y genéticos asociados al cáncer de mama en féminas del área de salud ¨28 de Septiembre¨. MEDISAN [revista en Internet]. 2011 [citado 30 Sep 2016];15(2):[aprox. 8p]. Disponible en: http://scieloprueba.sld.cu/scielo.php?script=sci_arttext&pid=S1029-30192011000200003&lng=es [Buscar en Google Scholar]
- Aguilar MJ, Neri M, Padilla CA, Pimentel ML, García A, Sánchez AM. Factores de riesgo como pronóstico de padecer cáncer de mama en un estado de México. Nutr Hosp [revista en Internet]. 2012 [citado 22 Sep 2016];27(5):[aprox. 6p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-16112012000500038&lng=es [Buscar en Google Scholar]
- Chlebowski RT, Anderson GL. Menopausal hormone therapy and breast cancer mortality: clinical implications. Ther Adv Drug Saf [revista en Internet]. 2015 [citado 2 Sep 2016];6(2):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4406918 [Buscar en Google Scholar]
- Antoine C, Ameye L, Paesmans M, Rozenberg S.. Systematic review about breast cancer incidence in relation to hormone replacement therapy use. Climacteric [revista en Internet]. 2014 [citado 12 Sep 2016];17(2):[aprox. 20p]. Disponible en: http://www.tandfonline.com/doi/full/10.3109/13697137.2013.829812 [Buscar en Google Scholar]
- Salagame U, Banks E, Sitas F, Canfell K. Menopausal hormone therapy use and breast cancer risk in Australia: findings from the New South Wales Cancer Lifestyle and EvAluation of Risk (CLEAR) study. Int J Cancer [revista en Internet]. 2016 [citado 2 Nov 2016];138(8):[aprox. 15p]. Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/ijc.29942/epdf [Buscar en Google Scholar]
- Rose DP, Gracheck PJ, Vona-Davis L. The Interactions of Obesity, Inflammation and Insulin Resistance in Breast Cancer. Cancers (Basel) [revista en Internet]. 2015 [citado 2 Sep 2016];7(4):[aprox. 20p]. Disponible en: http://www.mdpi.com/2072-6694/7/4/0883/htm [Buscar en Google Scholar]
- Boonyaratanakornkit V, Pateetin P. The role of ovarian sex steroids in metabolic homeostasis, obesity, and postmenopausal breast cancer: molecular mechanisms and therapeutic implications. Biomed Res Int [revista en Internet]. 2015 [citado 2 Sep 2016]; :[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4383469 [Buscar en Google Scholar]
- Harvie M, Howell A, Evans DG. Can diet and lifestyle prevent breast cancer: what is the evidence?. Am Soc Clin Oncol Educ Book [revista en Internet]. 2015 [citado 2 Sep 2016];35(1):[aprox. 8p]. Disponible en: http://meetinglibrary.asco.org/content/11500066-156 [Buscar en Google Scholar]
- Torres L. Superación profesional: una alternativa para el diagnóstico precoz del cáncer de mama. Medisur [revista en Internet]. 2012 [citado 12 Dic 2015];10(2):[aprox. 10p]. Disponible en: http://medisur.sld.cu/index.php/medisur/article/view/1980/920 [Buscar en Google Scholar]
- Osborne C, Wilson P, Tripathy D. Oncogenes and tumor suppressor genes in breast cancer: potential diagnostic and therapeutic applications. Oncologist [revista en Internet]. 2004 [citado 2 Nov 2015];9(4):[aprox. 15p]. Disponible en: http://theoncologist.alphamedpress.org/content/9/4/361.long [Buscar en Google Scholar]
- Padrón J, Padrón L, Padrón L, Morejón AF, Benet M. Comportamiento del diagnóstico precoz del cáncer de mama y cérvicouterino en el municipio Cienfuegos. Finlay [revista en Internet]. 2013 [citado 14 Mar 2016];3(2):[aprox. 7p]. Disponible en: http://revfinlay.sld.cu/index.php/finlay/article/view/187 [Buscar en Google Scholar]
- Torres L, Iglesias M, Zerquera C. Repercusión de la implementación del Diplomado de Mastología en la provincia de Cienfuegos. Medisur [revista en Internet]. 2011 [citado 14 Mar 2016];9(5):[aprox. 5p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1727-897X2011000500005&lng=es [Buscar en Google Scholar]
- Migliaccio I, Malorni L, Hart CD, Guarducci C, Di Leo A. Endocrine therapy considerations in postmenopausal patients with hormone receptor positive, human epidermal growth factor receptor type 2 negative advanced breast cancers. BMC Med [revista en Internet]. 2015 [citado 2 Sep 2016];13(1):[aprox. 13p]. Disponible en: http://www.biomedcentral.com/1741-7015/13/46 [Buscar en Google Scholar]
- Latasa P, Gandarillas AM, Ordobas M. Tendencias y desigualdades sociales en el cribado de cáncer de cérvix y cáncer de mama en la Comunidad de Madrid durante el periodo 1995-2010 a partir del Sistema de Vigilancia de Factores de Riesgo de enfermedades no transmisibles (SIVFRENT-A). Anales Sis San Navarra [revista en Internet]. 2015 [citado 9 Sep 2016];38(1):[aprox. 10p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1137-66272015000100003&lng=es [Buscar en Google Scholar]
- Vich P, Brusint B, Álvarez C, Cuadrado C, Díaz N, Redondo E. Actualización del cáncer de mama en Atención Primaria (I/V). Semergen [revista en Internet]. 2014 [citado 2 Nov 2015];40(6):[aprox. 5p]. Disponible en: http://dx.doi.org/10.1016/j.semerg.2014.02.012 [Buscar en Google Scholar]
- Kamińska M, Ciszewski T, Łopacka-Szatan K, Miotła P, Starosławska E. Breast cancer risk factors. Prz Menopauzalny [revista en Internet]. 2015 [citado 2 Sep 2016];14(3):[aprox. 7p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4612558 [Buscar en Google Scholar]
- Dieterich M, Stubert J, Reimer T, Erickson N, Berling A. Influence of lifestyle factors on breast cancer risk. Breast Care (Basel) [revista en Internet]. 2014 [citado 2 Nov 2015];9(6):[aprox. 7p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4317679 [Buscar en Google Scholar]
- Murria R, Palanca S, de Juan I, Egoavil C, Alenda C, García Z, et al. Methylation of tumor suppressor genes is related with copy number aberrations in breast cancer. Am J Cancer Res [revista en Internet]. 2014 [citado 2 Sep 2016];5(1):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4300703 [Buscar en Google Scholar]
- Miguel PE, Almaguer A, Ponce de León D, Sales H, Pérez H. El cáncer una enfermedad genética. CCM [revista en Internet]. 2007 [citado 2 Nov 2015];11(3):[aprox. 8p]. Disponible en: http://www.cocmed.sld.cu/no113/n113rev1.htm [Buscar en Google Scholar]
- Weitzel JN. The Genetics of Breast Cancer. What the Surgical Oncologist Needs to Know. Surg Oncol Clin N Am [revista en Internet]. 2015 [citado 2 Sep 2016];24(4):[aprox. 20p]. Disponible en: http://dx.doi.org/10.1016/j.soc.2015.06.011 [Buscar en Google Scholar]
- Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: new hereditary breast cancer susceptibility genes. Cancer Treat Rev [revista en Internet]. 2015 [citado 2 Nov 2016];41(1):[aprox. 7p]. Disponible en: http://dx.doi.org/10.1016/j.ctrv.2014.10.008 [Buscar en Google Scholar]
- Maxwell KN, Nathanson KL. Common breast cancer risk variants in the post-COGS era: a comprehensive review. Breast Cancer Res [revista en Internet]. 2013 [citado 2 Nov 2015];15(6):[aprox. 3p]. Disponible en: http://breast-cancer-research.com/content/15/6/212 [Buscar en Google Scholar]
- Davis NM, Sokolosky M, Stadelman K, Abrams SL, Libra M, Candido S, et al. Deregulation of the EGFR/PI3K/PTEN/Akt/mTORC1 pathway in breast cancer: possibilities for therapeutic intervention. Oncotarget [revista en Internet]. 2014 [citado 2 Nov 2015];5(13):[aprox. 20p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4148087 [Buscar en Google Scholar]
- Vogt PK. Retroviral Oncogenes: A Historical Primer. Nat Rev Cancer [revista en Internet]. 2012 [citado 2 Nov 2015];12(9):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3428493 [Buscar en Google Scholar]
- Shortt J, Johnstone RW. Oncogenes in Cell Survival and Cell Death. Cold Spring Harb Perspect Biol [revista en Internet]. 2012 [citado 2 Nov 2015];4(12):[aprox. 8p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3504432 [Buscar en Google Scholar]
- Bagci O, Kurtgöz S. Amplification of Cellular Oncogenes in Solid Tumors. N Am J Med Sci [revista en Internet]. 2015 [citado 2 Sep 2016];7(8):[aprox. 7p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4561439 [Buscar en Google Scholar]
- Omenn GS, Guan Y, Menon R. A new class of protein cancer biomarker candidates: differentially expressed splice variants of ERBB2 (HER2/neu) and ERBB1 (EGFR) in breast cancer cell lines. J Proteomics [revista en Internet]. 2014 [citado 2 Sep 2016];107(1):[aprox. 9p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4123867 [Buscar en Google Scholar]
- Vici P, Pizzuti L, Natoli C, Gamucci T, Di Lauro L, Barba M, et al. Triple positive breast cancer: a distinct subtype?. Cancer Treat Rev [revista en Internet]. 2015 [citado 2 Sep 2016];41(2):[aprox. 7p]. Disponible en: http://dx.doi.org/10.1016/j.ctrv.2014.12.005 [Buscar en Google Scholar]
- Hosford SR, Miller TW. Clinical potential of novel therapeutic targets in breast cancer: CDK4/6, Src, JAK/STAT, PARP, HDAC, and PI3K/AKT/mTOR pathways. Pharmgenomics Pers Med [revista en Internet]. 2014 [citado 2 Nov 2015];7(1):[aprox. 12p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4157397 [Buscar en Google Scholar]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature [revista en Internet]. 2012 [citado 2 Nov 2015];490(7418):[aprox. 9p]. Disponible en: http://www.nature.com/nature/journal/v490/n7418/full/nature11412.html [Buscar en Google Scholar]
- He X, Xiang H, Zong X, Yan X, Yu Y, Liu G, et al. CDK2-AP1 inhibits growth of breast cancer cells by regulating cell cycle and increasing docetaxel sensitivity in vivo and in vitro. Cancer Cell Int [revista en Internet]. 2014 [citado 2 Nov 2015];14(1):[aprox. 5p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4279590 [Buscar en Google Scholar]
- Ullah A, Mahjabeen I, Kayani MA. Genetic polymorphisms in cell cycle regulatory genes CCND1 and CDK4 are associated with susceptibility to breast cancer. J BUON [revista en Internet]. 2015 [citado 2 Sep 2016];20(4):[aprox. 8p]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/26416047 [Buscar en Google Scholar]
- Chen BJ, Wu YL, Tanaka Y, Zhang W. Small molecules targeting c-Myc oncogene: promising anti-cancer therapeutics. Int J Biol Sci [revista en Internet]. 2014 [citado 2 Nov 2015];10(10):[aprox. 12p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4202025 [Buscar en Google Scholar]
- Dey N, Leyland B, De P. MYC-xing it up with PIK3CA mutation and resistance to PI3K inhibitors: summit of two giants in breast cancers. Am J Cancer Res [revista en Internet]. 2014 [citado 2 Nov 2015];5(1):[aprox. 19p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4300701 [Buscar en Google Scholar]
- Minuti G, Landi L. MET deregulation in breast cancer. Ann Transl Med [revista en Internet]. 2015 [citado 2 Sep 2016];3(13):[aprox. 3p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4543340 [Buscar en Google Scholar]
- Graveel CR, Tolbert D, Woude GV. MET: A Critical Player in Tumorigenesis and Therapeutic Target. Cold Spring Harb Perspect Biol [revista en Internet]. 2013 [citado 2 Nov 2015];5(7):[aprox. 9p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3685898 [Buscar en Google Scholar]
- Graveel CR, DeGroot JD, Su Y, Koeman J, Dykema K, Leung S, et al. Met induces diverse mammary carcinomas in mice and is associated with human basal breast cancer. Proc Natl Acad Sci USA [revista en Internet]. 2009 [citado 2 Nov 2015];106(31):[aprox. 5p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2722304 [Buscar en Google Scholar]
- Ho-Yen CM, Jones JL, Kermorgant S. The clinical and functional significance of c-Met in breast cancer: a review. Breast Cancer Res [revista en Internet]. 2015 [citado 2 Sep 2016];17(1):[aprox. 2p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4389345 [Buscar en Google Scholar]
- Yang ZY, Di MY, Yuan JQ, Shen WX, Zheng DY, Chen JZ, et al. The prognostic value of phosphorylated Akt in breast cancer: a systematic review. Sci Rep [revista en Internet]. 2015 [citado 2 Sep 2016];5(1):[aprox. 6p]. Disponible en: http://www.nature.com/articles/srep07758 [Buscar en Google Scholar]
- Faes S, Dormond O. PI3K and AKT: Unfaithful Partners in Cancer. Int J Mol Sci [revista en Internet]. 2015 [citado 2 Sep 2016];16(9):[aprox. 15p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4613246 [Buscar en Google Scholar]
- Scalia J, Colins BL, Penson RT, Dizon DS. Breast Cancer Risk Assessment: Moving Beyond BRCA 1 and 2. Semin Radiat Oncol [revista en Internet]. 2016 [citado 2 Nov 2016];26(1):[aprox. 5p]. Disponible en: http://www.semradonc.com/article/S1053-4296(15)00095-8/pdf [Buscar en Google Scholar]
- Agarwal R, Liebe S, Turski ML, Vidwans SJ, Janku F, Garrido I, et al. Targeted therapy for hereditary cancer syndromes: hereditary breast and ovarian cancer syndrome, Lynch syndrome, familial adenomatous polyposis, and Li-Fraumeni syndrome. Discov Med [revista en Internet]. 2014 [citado 2 Nov 2015];18(101):[aprox. 8p]. Disponible en: http://www.discoverymedicine.com/Rishi-Agarwal/2014/12/targeted-therapy-of-hereditary-cancer-syndromes-hereditary-breast-and-ovarian-cancer-syndrome-lynch-syndrome-familial-adenomatous-polyposis-and-li-fraumeni-syndrome [Buscar en Google Scholar]
- Ortigoza RI, Miguel PE, Machín D. Variantes genéticas en el cáncer de mama. CCM [revista en Internet]. 2012 [citado 2 Nov 2015];16(4):[aprox. 6p]. Disponible en: http://revcocmed.sld.cu/index.php/cocmed/article/view/737/230 [Buscar en Google Scholar]
- Walerych D, Napoli M, Collavin L, Del Sal G. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis [revista en Internet]. 2012 [citado 2 Nov 2015];33(11):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3483014 [Buscar en Google Scholar]
- Rondeau S, Vacher S, De Koning L, Briaux A, Schnitzler A, Chemlali W, et al. ATM has a major role in the double-strand break repair pathway dysregulation in sporadic breast carcinomas and is an independent prognostic marker at both mRNA and protein levels. Br J Cancer [revista en Internet]. 2015 [citado 2 Sep 2016];112(6):[aprox. 7p]. Disponible en: http://www.nature.com/bjc/journal/v112/n6/full/bjc201560a.html [Buscar en Google Scholar]
- Rhodes LV, Tate CR, Hoang VT, Burks HE, Gilliam D, Martin EC, et al. Regulation of triple-negative breast cancer cell metastasis by the tumor-suppressor liver kinase B1. Oncogenesis [revista en Internet]. 2015 [citado 2 Sep 2016];4(1):[aprox. 9p]. Disponible en: http://www.nature.com/oncsis/journal/v4/n10/full/oncsis201527a.html [Buscar en Google Scholar]
- Witkiewicz AK, Knudsen ES. Retinoblastoma tumor suppressor pathway in breast cancer: prognosis, precision medicine, and therapeutic interventions. Breast Cancer Res [revista en Internet]. 2014 [citado 2 Nov 2015];16(3):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4076637 [Buscar en Google Scholar]
- Andrews JL, Kim AC, Hens JR. The role and function of cadherins in the mammary gland. Breast Cancer Res [revista en Internet]. 2012 [citado 2 Nov 2015];14(1):[aprox. 2p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3496113 [Buscar en Google Scholar]
- Asiaf A, Ahmad ST, Aziz SA, Malik AA, Rasool Z, Masood A, Zargar MA. Loss of expression and aberrant methylation of the CDH1 (E-cadherin) gene in breast cancer patients from Kashmir. Asian Pac J Cancer Prev [revista en Internet]. 2014 [citado 2 Nov 2015];15(15):[aprox. 6p]. Disponible en: http://www.apocpcontrol.org/paper_file/issue_abs/Volume15_No15/6397-6403 6.4 Asia Asiaf.pdf [Buscar en Google Scholar]
- Repetto O, De Paoli P, De Re V, Canzonieri V, Cannizzaro R. Levels of soluble E-cadherin in breast, gastric, and colorectal cancers. Biomed Res Int [revista en Internet]. 2014 [citado 2 Nov 2015];2014(1):[aprox. 10p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4182303 [Buscar en Google Scholar]
- MacNeil AJ, Jiao SC, McEachern LA, Yang YJ, Dennis A, Yu H, et al. MAPK kinase 3 is a tumor suppressor with reduced copy number in breast cancer. Cancer Res [revista en Internet]. 2014 [citado 2 Feb 2016];74(1):[aprox. 10p]. Disponible en: http://cancerres.aacrjournals.org/content/74/1/162.long [Buscar en Google Scholar]
- García C, Cortijo S, Cañamares I, Goyache MP, Ferrari JM. Lapatinib en combinación con trastuzumab en el tratamiento del cáncer de mama metastásico HER-2 positivo: experiencia de uso. Farm Hosp [revista en Internet]. 2014 [citado 14 Dic 2015];38(2):[aprox. 5p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S1130-63432014000200009&lng=es [Buscar en Google Scholar]
- Germain D. Estrogen Carcinogenesis in Breast Cancer. Endocrinol Metab Clin North Am [revista en Internet]. 2011 [citado 12 Nov 2015];40(3):[aprox. 11p]. Disponible en: https://www.clinicalkey.es/ui/service/content/url?eid=1-s2.0-S0889852911000582 [Buscar en Google Scholar]
- Stuckey AR, Onstad MA. Hereditary breast cancer: an update on risk assessment and genetic testing in 2015. Am J Obstet Gynecol [revista en Internet]. 2015 [citado 2 Sep 2016];213(2):[aprox. 6p]. Disponible en: http://dx.doi.org/10.1016/j.ajog.2015.03.003 [Buscar en Google Scholar]
- Tavtigian SV, Chenevix G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark Med [revista en Internet]. 2014 [citado 2 Nov 2015];8(4):[aprox. 15p]. Disponible en: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC4336165 [Buscar en Google Scholar]
- Chen J, Weiss WA. Alternative splicing in cancer: implications for biology and therapy. Oncogene [revista en Internet]. 2015 [citado 2 Sep 2016];34(1):[aprox. 13p]. Disponible en: http://www.nature.com/onc/journal/v34/n1/full/onc2013570a.html [Buscar en Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet [revista en Internet]. 2007 [citado 23 Mar 2016];8(10):[aprox. 13p]. Disponible en: http://www.nature.com/nrg/journal/v8/n10/full/nrg2159.html [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129