
Presentaciones de casos
Iniencefalia apertus fetal. Reporte de un caso y revisión de la literatura
Fetal Apertus Iniencephaly. Case Report and Literature Review

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2026-02-21 19:40:40
Aprobado: 2026-02-23 12:04:58
Correspondencia: Lilian Rachel Vila Ferrán. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. lilian.vila@gal.sld.cu
RESUMEN
Palabras clave: malformación; tubo neural; patología forense
ABSTRACT
Key words: malformation; neural tube; forensic pathology
INTRODUCCIÓN
La iniencefalia es la herniación de la masa cerebral y las meninges a través de un defecto de fusión de las vértebras iniciales de la columna. Por otra parte, suele faltar además parte de la escama del occipital con agrandamiento del agujero occipital. De ahí que muchos autores incluyan ya este proceso dentro de los encefaloceles. También se define como una gran hiperextensión de la cabeza fetal, con el occipucio fundiéndose con la columna vertebral. Es una malformación rara, de pronóstico grave, que implica principalmente al occipucio (del griego: inion) y al cuello. Pertenece al espectro de anomalías de cierre del tubo neural, de ahí que participe de un determinismo multifactorial.(1,2,3,4,5,6)
La iniencefalia raramente se encuentra asociada a cromosomopatías. Se han reportado monosomía del X, trisomía 2 y trisomía 13 en mosaico.(4,6,7) Se presenta con mayor frecuencia en fetos del sexo femenino, habitualmente esporádica, no obstante, las cifras varían según los autores: (sex ratio de 1/3 a 1/9), como es este caso del sexo masculino. La hipótesis de una penetrancia incompleta y/o de una mortalidad masculina acrecentada se han insinuado, pero no confirmado.(1,3,4,8,9)
Se ha reportado que tiene relación con la consanguinidad entre los padres, también se atribuye a la relación con sífilis materna, la ingestión de tetraciclinas, citrato de clomifeno y sedantes por parte de la madre, aunque no existen evidencias definitivas.(3,6,7) En este caso no se recogieron ninguno de estos antecedentes.
Las causas de la iniencefalia son aún desconocidas(3,6,10) pero algunos autores plantean una anomalía del desarrollo de la base del cráneo y de la columna vertebral cardiotorácica, anterior al cierre del neuróporo cefálico hacia los 24-30 días de la gestación (el neuróporo caudal alrededor de los 31 días). El cierre del neuróporo rostral o cerebral es básicamente bidireccional, es decir, se produce en ambas direcciones, rostral y caudal. El neuroporo rostral se sitúa probablemente en la futura placa comisural, en medio de la lámina terminal embrionaria. Los esclerotomas paravertebrales, de origen mesodérmico, se diferencian de manera precoz en una masa ventral (cuerpos vertebrales y pedículos) y una masa dorsal (arcos posteriores y bóveda craneana. La hipoplasia de las masas mesodérmicas ventrales y/o dorsales determinarían la malformación característica de la iniencefalia, con un defecto complejo que permite la comunicación directa entre la fosa posterior y la porción cervical del canal medular.(1,6) En el caso presentado abarcaba hasta la región dorsal.
Se puede presentar en un contexto familiar de anomalía de cierre otro segmento del tubo neural, con un riesgo de recurrencia estimado de entre el 1 y el 5 %. Esta malformación puede integrarse en diferentes síndromes o en el fenotipo de ciertas aberraciones cromosómicas (trisomía 18, triploidía, monosomía X) y justifica la indicación sistemática del cariotipo. No se ha podido establecer ninguna anomalía cromosómica específicamente asociada o ligada a la iniencefalia; esto confirma la hipótesis de un determinismo multifactorial, genético y ambiental, de la malformación y de los defectos de cierre del tubo neural en general.(1,2)
La iniencefalia es un padecimiento raro, su frecuencia aún no se ha evaluado correctamente, ya que hasta la actualidad solo se han reportado 200 casos en la literatura científica, aunque parece ser que su incidencia global está estimada entre 1 de cada 1.000 y 1 de cada 100.000 nacimientos según diferentes países y regiones. Su distribución geográfica es heterogénea, posiblemente superponible a la de la anencefalia, por lo que hay mecanismos fisiopatogénicos multifactoriales comunes.(1,2,3,4,5,6)
La revisión de la literatura pretende describir la etiología, incidencia, características, métodos de diagnóstico y pronóstico de la iniencefalia, debido a la baja frecuencia con que esta entidad se presenta para su mejor estudio y comprensión y así elevar la calidad de la atención ante estos raros y complejos casos.
Este reporte subraya la importancia del diagnóstico ecográfico temprano y el rol del estudio postmortem como herramienta fundamental para la confirmación diagnóstica y la clasificación precisa de esta compleja anomalía.
PRESENTACIÓN DEL CASO
Se presenta el caso de una gestante de 32 años de edad, primigesta, sin antecedentes patológicos personales ni familiares de interés, ni exposición a factores teratógenos. La paciente fue remitida con 20,5 semanas de amenorrea al Servicio de Genética Médica de Cienfuegos. En la exploración ecográfica se observó feto masculino acorde a su edad gestacional con orientación de la cara hacia arriba, ausencia de cuello, defecto a nivel de vértebras cervicales y dorsales con salida de meninges y médula espinal, ausencia de cuerpo calloso, pie derecho con la planta girada hacia dentro y en punta.
El diagnóstico planteado por el Servicio de Genética Médica fue: iniencefalia con holoprosencefalia alobar, pie varoequino derecho. Se le brindó asesoramiento genético a la pareja y esta decidió la interrupción del embarazo, que se realizó a las 21 semanas. Se obtuvo cadáver de sexo masculino, de color de piel blanca, al que se le realizó necropsia clínica en el Hospital General Universitario Dr. Gustavo Aldereguía Lima de Cienfuegos.
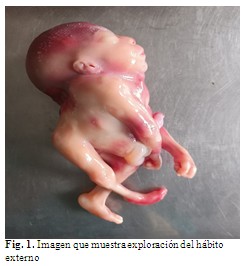
A la exploración del hábito externo se observó desproporción entre la talla y la brazada, fue esta última la mayor, hiperextensión del cuello que llevó a retroflexión forzada de la cabeza, con unión de la porción occipital a la región cervical y torácica de la columna, con continuidad de tejidos blandos a la espalda y ausencia de cuello, orejas de implantación baja, mandíbula pequeña, atrofia del músculo estriado esquelético de la pierna derecha, pie varoequino derecho con desproporción del tamaño entre ambos pies. (Fig. 1).
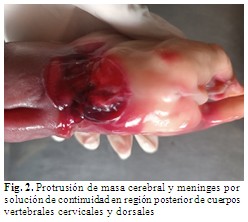
Se observó protrusión de masa cerebral, médula espinal y meninges por solución de continuidad de 4 cm de diámetro en región posterior de cuerpos vertebrales cervicales y dorsales. (Fig. 2).
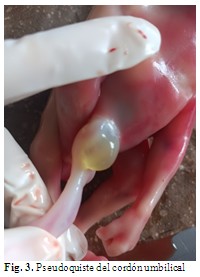
Se observó acúmulo de líquido claro, semejante a un quiste de 1 cm de diámetro, renitente al tacto, traslúcido, superficie lisa, en la base de implantación abdominal del cordón umbilical. (Fig. 3).
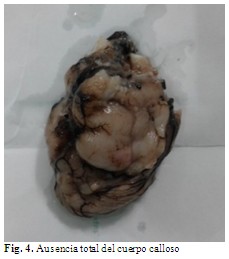
A la disección por aparatos se encontró aumento en el tamaño de la fontanela anterior y ausencia total del cuerpo calloso y ensanchamiento del agujero occipital. (Fig. 4).
En el estudio anatomopatológico se determinó: iniencefalia apertus con holoprosencefalia alobar, fontanela anterior muy aumentada en tamaño, implantación baja de las orejas, micrognatia, atrofia del músculo estriado esquelético de la pierna derecha, pie varoequino derecho con desproporción del tamaño entre ambos pies y pseudoquiste del cordón umbilical.
DISCUSIÓN
En 1897 Lewis describió dos tipos de iniencefalia: abierta (apertus), cuando se presenta con encefalocele y cerrada (clausus), cuando no existe malformación del occipital y el cerebro está presente.(3,5 6,7)
También se ha clasificado como iniencefalia simple (apertus o clausus) e iniencefalia con anencefalia (anencefalia con retroflexión espinal).(6)
La presentación macroscópica y ecográficas del feto iniencefálico son características. Posee una tríada diagnóstica definida por Ballantyne en 1904: anomalía del desarrollo de la base del cráneo con ensanchamiento del orificio occipital, disrafismo cervical e hiperextensión de la cabeza con hiperlordosis cervicotorácica extrema y ausencia característica del cuello.
La masa facial aparece aplastada, orientada hacia arriba, con los ojos exorbitados. La implantación de los pabellones auditivos es baja. Existe bóveda craneana y cuero cabelludo, el límite de implantación de los cabellos alcanza las escápulas. La columna vertebral es corta debido a la hiperextensión de la región cervicotorácica y la lordosis exagerada. Se observa también una espina bífida acompañanda de iniencefalia en el 75 % de los casos. Las vértebras cervicales y torácicas altas son anormales, poco numerosas, a menudo fusionadas con los arcos incompletamente cerrados.(1,2,4) El caso presentado tiene esta triada diagnóstica.
Entre el 50 y el 85 % de los casos se asocian con otras malformaciones. Puede tratarse de malformaciones del SNC, anomalías faciales u otras malformaciones viscerales. Dentro de las más frecuentes se encuentran la anencefalia, onfalocele, encefalocele, ciclopia, hernia diafragmática, atresias intestinales, mielomeningocele, cardiopatías, holoprosencefalia, queilopalatosquisis, hidronefrosis, fisura palatina, arteria umbilical única y anomalías histológicas como riñones multiquísticos.(1,2,3,4,6,7,8,9) Este caso además presentó holoprosencefalia y encefalocele.
Se informa que en el 84 % de los casos se asocian otras malformaciones como ausencia de la mandíbula, labio y paladar hendido, hidrocefalia, microcefalia, polimicrogiria, agenesia del vermis cerebeloso y quistes en el cerebelo.(3,7)
Las observaciones ecográficas características de la iniencefalia son la extensión mayor de la cabeza, la continuidad de la cabeza y el tronco, sin cuello visible, el acortamiento del raquis y vértebras cervicales mal individualizadas (número y forma), junto con la falta de cierre del arco posterior. El diámetro biparietal puede estar aumentado si existe una dilatación ventricular. De manera eventual se pueden encontrar anomalías costales como el aspecto delgado, bifidad. La ecografía confirma el hidramnios y el retraso del crecimiento intrauterino y participa en el pronóstico de gravedad en la búsqueda de malformaciones asociadas.(1,2,3,4,6)
Ecográficamente, la imagen en un primer momento es muy semejante a la propia del encefalocele occipital (la profusión del cerebro y las meninges a través de un agujero anormal del cráneo produciéndose de forma más frecuente en la región occipital o en la frontal). Se trata de una tumoración ecopositiva de tamaño variable adherida al occipucio. Su interior puede ser quístico (lleno de líquido cefalorraquídeo) o semisólido (con estructuras encefálicas en su interior). Pero en los cortes transversales cuidadosos se observa un arco vertebral posterior en forma de «V» en la región alta del raquis. Este signo es la clave de su diagnóstico diferencial con el encefalocele.(1)
La resonancia magnética puede completar de manera eventual la exploración ecográfica de una anomalía del SNC.(1,10,11)
Las concentraciones de alfafetoproteína en el líquido amniótico pueden estar aumentadas, en consonancia con el defecto de cierre del tubo neural, es en la actualidad, la alfafetoproteína en suero materno el método de cribado más importante para detectar defectos del tubo neural.(1,2,6,9)
En los casos de iniencefalia es conveniente la realización de cariotipo con bandas G de alta resolución (no realizada en este caso), de muestra obtenida por biopsia de vellosidad coriónica, amniocentesis, cordocentesis o en el recién nacido, para descartar alteraciones cromosómicas numéricas o estructurales.(2,6)
El diagnóstico prenatal de iniencefalia y anomalías asociadas establecería el pronóstico del feto, daría la posibilidad de la interrupción temprana del embarazo por decisión de la pareja, permitiría la preparación temprana del equipo médico para la atención de la madre, el parto y el feto, con el fin de evitar una cesárea. Este diagnóstico temprano también ayudaría a sensibilizar a la familia acerca de la importancia de la realización de autopsia fetal que incluya radiografía y resonancia magnética para delimitar los defectos óseos.(1,2,6)
Dentro de los diagnósticos diferenciales de este padecimiento se incluyen: el síndrome de Klipper-Feil, la anencefalia con retroversión espinal, el mielomeningocele cervical, la malformación de Dandy-Walker(1,3) y la disostosis espondilocostal o síndrome de Jarcho-Levin.(6)
La anencefalia se caracteriza por una ausencia completa o parcial del neurocraneo, y en la iniencefalia, la cabeza retroflexionada está completamente cubierta de piel. En cuanto al síndrome de Klippel-Feil y la iniencefalia, hay anomalías segmentarias cervicales en ambos, pero la hiperextensión no se observa en el síndrome de Klippel-Feil. Además, este síndrome no es mortal y puede tratarse con cirugía.(10)
El pronóstico es grave, y según la mayor parte de los autores, la supervivencia se torna imposible.(1,2, 3,5,6,11)
Con el nacimiento, el feto puede nacer muerto o sobrevivir horas o días. La iniencefalia se complica, a veces, por la hipertensión arterial materna, y frecuentemente por una presentación de nalgas, causante de una distocia que agrava aún más la malformación cervicooccipital irreductible.(1)
Este caso subraya la importancia del diagnóstico ecográfico temprano y el rol del estudio postmortem como herramienta fundamental para la confirmación diagnóstica y la clasificación precisa de esta compleja anomalía, lo que permite un adecuado estudio genético y asesoramiento sobre el riesgo de recurrencia.
Conflicto de intereses:
Las autoras declaran la no existencia de conflictos de intereses relacionados con el estudio.
Roles de autoría:
1. Conceptualización: Lilian Rachel Vila Ferrán, Tania Bernal Medina, Leydi María Sosa Águila.
2. Visualización: Lilian Rachel Vila Ferrán.
2. Redacción de borrador original: Lilian Rachel Vila Ferrán, Tania Bernal Medina, Leydi María Sosa Águila.
3. Redacción, revisión y edición: Lilian Rachel Vila Ferrán.
REFERENCIAS BIBLIOGRÁFICAS
1. Tejerizo A, Marinob M, Belloso M, Villalba A, González SP, Ruiz MA, et al. Iniencefalia. Rev Clin Inves Ginecol Obstec[Internet]. 2006[citado 28/1/2026];33(4):[aprox. 9p.]. Disponible en: https://www.elsevier.es/es-revista-clinica-e-investigacion-ginecologia-obstetricia-7-articulo-iniencefalia-13090044.
2. Saldarriaga W, Ruiz FA, Isaza C. Iniencefalia: primer caso reportado en Colombia y revisión de la literatura. Rev Colomb Obstet Ginecol[Internet]. 2011[citado 14/2/2026];62(4):[aprox. 5p.]. Disponible en: https://www.scielo.org.co/pdf/rcog/v62n4/v62n4a09.pdf.
3. Ministerio de Salud Brasil. Guía Práctica: Diagnóstico de anomalías congénitas en el prenatal y al nacimiento[Internet]. Brasilia:MSB;2023[citado 5/3/2026]. Disponible en: https://bvsms.saude.gov.br/bvs/publicacoes/guia_diagnostico_anomalias_congenitas_espanhol.pdf.
4. Sociedad Internacional de Ultrasonido en Obstetricia y Ginecología. Iniencefalia[Internet]. Reino Unido:SIUOG;2022[citado 24/3/2026]. Disponible en: https://www-isuog-org.translate.goog/clinical-resources/patient-information-series/patient-information-pregnancy-conditions/brain/iniencephaly.html?_x_tr_sl=en&_x_tr_tl=es&_x_tr_hl=es&_x_tr_pto=tc.
5. Sabharwal G, Agarwal M. Antenatal ultrasound diagnosis of Iniencephaly Apertus. International Society for Gynecologic Endoscopy[Internet]. 2021[citado 28/12/2025]; 2(1):[aprox. 10p.]. Disponible en: https://www.isge.org/wp-content/uploads/2021/03/TheTrocar-Vol2-Iss1_AntenatalUltrasoundDiagnosis.pdf.
6. Torres D, Reyna E, Rondon M. Diagnóstico prenatal de iniencefalia. Reporte de caso. Rev Obstet Ginecol Venez[Internet]. 2021[citado 16/4/2026];81(3):[aprox. 5p.]. Disponible en: https://saber.ucv.ve/ojs/index.php/rev_ogv/article/view/23191/144814489460.
7. National Library of Medicine. Trisomía 13[Internet]. Bethesda:MedlinePlus;2025[citado 11/9/2026]. Disponible en: https://medlineplus.gov/spanish/ency/article/001660.htm.
8. Copp AJ, Greene DE. Neural tube defects: disorders of neurulation and related embryonic processes. Wiley Interdiscip Rev Dev Biol. 2012;2(2):213-27.
9. Shaikh F, Sanadhya M, Kaleem S, Verma T, Jayaraj RL, Ahmad F. Evaluación crítica de los defectos del tubo neural y sus complejidades. Pediatr Neonatol[Internet]. 2025[citado 14/1/2026];66(6):[aprox. 10p.]. Disponible en: https://www-sciencedirect-com.translate.goog/science/article/pii/S1875957225000828?_x_tr_sl=en&_x_tr_tl=es&_x_tr_hl=es&_x_tr_pto=tc.
10. Arega BN, Endalew SD, Hailu DM. A Rare Case of Fetal Neural Tube Defect. Iniencephaly Clausus. AJP Rep. 2024;14(4):281-3.
11. Minchola JL, Zamora VE, Lazarte CI. Prenatal imaging diagnosis of iniencephaly apertus associated with heterotaxy syndrome, alobar holoprosencephaly and myelomeningocele: a case report. AJOG Glob Rep. 2025;5(3):100539.
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129