
Presentaciones de casos
Delineación dismorfológica del síndrome orofaciodigital Tipo I. Presentación de un caso y revisión de la literatura
Dysmorphological Delineation of Orofaciodigital Syndrome Type I. Presentation of a Case and Literature Review

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-06-20 10:04:53
Aprobado: 2024-06-26 00:02:38
Correspondencia: Noel Taboada Lugo. Facultad de Estomatología. Universidad de Ciencias Médicas de Villa Clara. Villa Clara. noeltl@infomed.sld.cu
RESUMEN
Palabras clave: trastornos del desarrollo infantil; discapacidad intelectual; reporte de casos
ABSTRACT
Key words: childhood development disorders; intellectual disability; case report
INTRODUCCIóN
Los síndromes orofaciodigitales (SOFD) son un grupo heterogéneo de trastornos del desarrollo, de los que se han descrito al menos, 18 variantes clínicas.(1) Como sugiere el nombre, los síndromes orofaciodigitales (SOFD) constituyen un grupo de alteraciones genéticas de origen monogénico que afectan principalmente el desarrollo de la cavidad oral: boca, lengua, dientes y mandíbula; la cara: cabeza, ojos y nariz y los dedos de las manos y los pies.(2,3)
La primera descripción de los SOFD fue realizada por Otto Mohr en 1941, mientras que el SOFD Tipo I (OMIM 605041) fue descrito por primera vez por Papillon-Leage y Psaume en 1954 y definido por Gorlin y Psaume en 1962, por lo que también se le conoce con el nombre de síndrome de Papillon-Leage Psaume.(2,4)
El SOFD Tipo I se transmite con herencia dominante ligada al X letal en varones hemicigóticos y presenta una gran expresividad variable inter e intrafamiliar. Entre un 70 y un 75 % de los casos son esporádicos y muchos de ellos son el resultado de variantes patogénicas de novo.(4,5) Tiene una incidencia entre 1: 50 000 a 250 000 nacidos vivos.(3,4)
Se debe a variantes de secuencias patogénicas en el gen OFD-1 (OMIM 311200) que consta de 23 exones, tiene locus génico en Xp22.3- p22.2 y codifica una proteína de 1011 aminoácidos, expresada en el centrosoma y en el cuerpo basal de los cilios primarios, por lo que se considera como una ciliopatía.(1,4)
Los síndromes muestran gran expresividad variable, lo que requiere el reconocimiento y el diagnóstico diferencial de los signos y síntomas clínicos que se presentan.
El diagnóstico clínico de sospecha del SOFD Tipo I se establece en la infancia por las dismorfias orofaciales y digitales o en adultos por asociarse a riñones poliquísticos. Se recomienda control anual de tensión arterial, función renal y ecografía renal porque en el 50 % o más de las mujeres aparecen riñones poliquísticos y puede ser la única manifestación.(1)
Los SOFD son el resultado del efecto pleiotrópico de una alteración morfogenética que afecta casi invariablemente a la boca, la cara y los dedos. Otros sistemas de órganos pueden estar involucrados, pero que definen tipos específicos de SOFD, por lo que el diagnóstico diferencial debe realizarse, en primer lugar, con los otros tipos de SOFD, de acuerdo a las diferentes características fenotípicas.
Aunque en todos los tipos de SOFD se constatan alteraciones dismorfológicas a nivel de la cavidad oral, la cara y los dedos, existen características fenotípicas distintivas en algunos de ellos que permiten diferenciarlo del SOFD Tipo I, además por los diferentes patrones de herencia monogénica.(6)
El diagnóstico diferencial se debe realizar también con la poliquistosis renal autosómica dominante, sobre todo en pacientes adultos, en los no se constata la presencia del patrón dismórfico orofaciodigital característico del SOFD.(1)
Su interés radica en presentar la delineación fenotípica de un síndrome poco frecuente, que puede manifestarse con una expresión clínica leve o asociado a importantes alteraciones dismorfológicas.
PRESENTACIóN DEL CASO
Se presenta el caso de un paciente de siete años de edad con antecedentes de retraso del desarrollo psicomotor que llevaba seguimiento en el Servicio de Cirugía Maxilofacial por presencia de fisura palatina, lengua multilobulada y múltiples frenillos gingivolabiales, para lo que se requirieron múltiples intervenciones quirúrgicas.
El paciente fue remitido al Servicio de Genética Clínica por presencia de patrón dismórfico craneofacial. Al examen dismorfológico se constató patrón dismórfico dado por frente amplia y prominente, pelo fino y ralo, hipotricosis con zonas alopécicas. (Fig. 1).
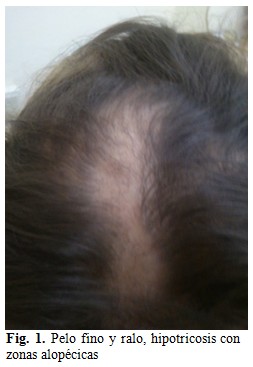
También se observó raíz nasal ancha con punta aplanada e hipoplasia de alas nasales, presencia de milium facial, a nivel de los ojos se observaba pliegue epicanto bilateral y pseudohipertelorismo por presencia de telecanto.
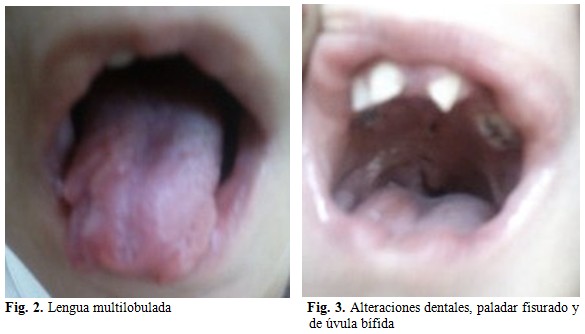
En la cavidad oral se constataron malposiciones dentarias severas, micrognatismo transversal, así como, anomalías de la forma dental (incisivos centrales cónicos o lobodontia) y de número dentarios (oligodoncia), necrosis pulpares, lengua multilobulada, fisura palatina y úvula bífida. (Figs. 2 y 3).
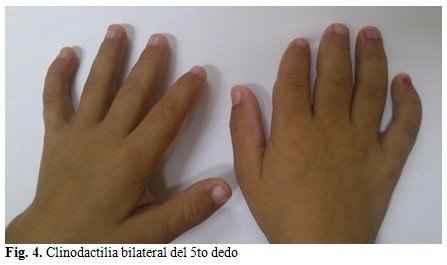
A nivel de las extremidades se observó clinodactilia bilateral del 5to dedo. (Fig. 4).
Se solicitó valoración por el Centro de Diagnóstico y Orientación (CDO) donde se diagnosticó la presencia de discapacidad intelectual moderada.
Se indicaron exámenes complementarios, entre ellos estudios de la orina (urocultivo, conteo de Addis, proteinuria de 24 horas y calciuria), estudios hematológicos y de hemoquímica de función renal, todos con resultados normales, así como ultrasonografía renal para descartar presencia de quistes renales, que no se identificaron en ninguno de los estudios sonográficos realizados en las consultas de seguimiento.
No se recogieron antecedentes familiares de enfermedades genéticas ni de defectos congénitos.
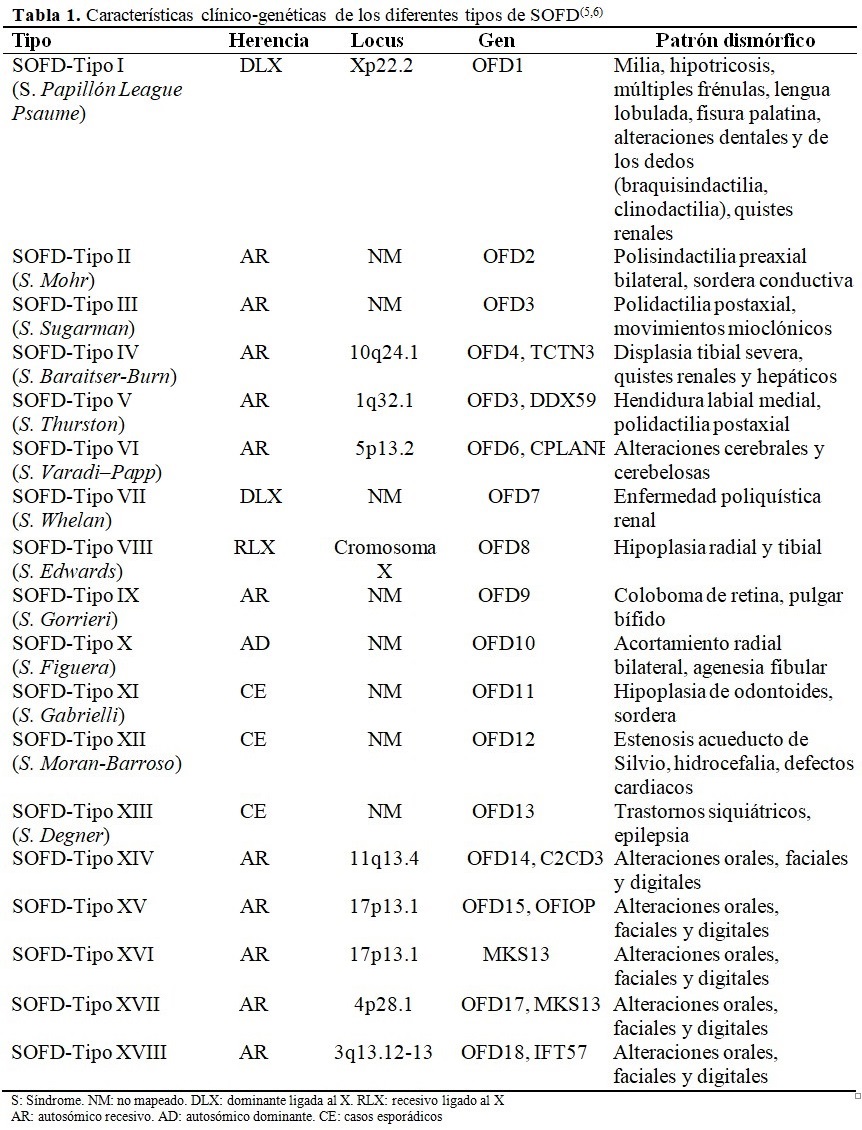
Dada las alteraciones dismorfológicas a nivel de la cara, región oral y de los dedos y ante la imposibilidad de realizar estudios moleculares, se planteó el diagnóstico de sospecha de SOFD Tipo 1, mediante la utilización del método clínico,(7) luego de realizar el diagnóstico diferencial con los otros tipos de SOFD. (Tabla 1).
Se obtuvo el consentimiento informado de la tutora de la paciente para la obtención de las fotografías y la publicación del reporte, de acuerdo a los principios éticos establecidos para estos casos.(8)
DISCUSIóN
El término "cilio primario" fue acuñado por Sergei Sorokin para describir un orgánulo que emana de la superficie celular de la mayoría de los tipos de células de mamíferos durante la detención del crecimiento. El cilio primario proporciona un medio para secuestrar el centriolo, por lo que la mayoría de las células que tienen cilios primarios son células diferenciadas no cíclicas o células madre en fase G0.
Cada vez, existen más pruebas que sugieren que el cilio primario es el coordinador clave durante el desarrollo y en la homeostasis tisular. Los cilios primarios también desempeñan un papel vital en la modulación de las vías de señalización celular. Por lo tanto, el cilio primario ayuda a orquestar procesos claves de desarrollo como la migración celular, la diferenciación celular, el control del ciclo celular, el plano de división celular y la apoptosis. Las vías de señalización moduladas a nivel del cuerpo basal del cilio primario son diversas y dependen del tipo de célula. La proteína SOFD es una de las proteínas asociadas con el cuerpo basal del cilio primario, que explica las manifestaciones fenotípicas del síndrome SOFD Tipo I, por efecto pleiotrópico del gen.(4)
La integridad funcional y estructural de los cilios influye en los procesos críticos de desarrollo cerebral, lo que explica la amplia gama de anomalías del neurodesarrollo en pacientes con ciliopatía. Se han identificado trastornos del espectro autista en una niña con SOFD Tipo I.(9)
Las ciliopatías incluyen afecciones más comunes, como la poliquistosis renal autosómica dominante y trastornos más raros, como el SOFD I y el síndrome de Joubert. La disfunción ciliar también puede dar lugar a trastornos que afectan a órganos individuales, como la retinosis pigmentaria.(10)
El manejo clínico de estos casos es multidisciplinario y depende de la severidad de la expresión fenotípica resultante de la variante de secuencia patogénica en el gen SOFD Tipo I.
Los pacientes diagnosticados con SOFD Tipo I presentan riesgo de desarrollar poliquistosis renal, por lo que deben mantenerse en observación con una cuidadosa evaluación morfológica y seguimiento bioquímico de la función renal.
Dado que la paciente no tenía un diagnóstico clínico de su condición médica se le brindó asesoramiento genético a la familia y se le orientó seguimiento regular por las diferentes especialidades médicas y estomatológicas, con las que se mantiene en seguimiento en la actualidad.
Es de vital importancia la presentación de la delineación fenotípica por lo poco frecuente de este síndrome que puede manifestarse con una expresión clínica leve o asociado a importantes alteraciones dismorfológicas.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Noel Taboada Lugo, Ana María Rodríguez Díaz, Tairí Borges García.
2. Curación de datos: Noel Taboada Lugo.
3. Análisis formal: Noel Taboada Lugo, Ana María Rodríguez Díaz.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: Noel Taboada Lugo, Ana María Rodríguez Díaz, Tairí Borges García.
6. Metodología: Noel Taboada Lugo.
7. Administración del proyecto: Noel Taboada Lugo.
8. Recursos: Noel Taboada Lugo, Ana María Rodríguez Díaz, Tairí Borges García.
9. Software: Noel Taboada Lugo, Ana María Rodríguez Díaz, Tairí Borges García.
10. Supervisión: Noel Taboada Lugo.
11. Validación: Noel Taboada Lugo.
12. Visualización: Noel Taboada Lugo, Ana María Rodríguez Díaz, Tairí Borges García.
13. Redacción – borrador original: Noel Taboada Lugo.
14. Redacción – revisión y edición: Ana María Rodríguez Díaz, Tairí Borges García.
REFERENCIAS BIBLIOGRÁFICAS
- Martínez V, Ortuño PP, Roca S, Rodríguez L, Galán I, Galbis L, et al. Síndrome orofaciodigital Tipo I: en el diagnóstico diferencial de la poliquistosis renal autosómica dominante, a próposito de 3 casos. Nefrol [Internet]. 2023 [citado 11 Sep 2024];43(2):[aprox. 13p]. Disponible en: https://www.revistanefrologia.com/es-pdf-S0211699521001417 [Buscar en Google Scholar]
- National Organization for Rare Disorders. Oral facial digital [Internet]. Washington, DC: NORD; 2021 [citado 12 Feb 2023]. Disponible en: https://rarediseases.org/rare-diseases/oral-facial-digital-syndrome/# [Buscar en Google Scholar]
- Nicklaus Children's Hospital. Oral Facial Digital Syndrome [Internet]. Florida: NCH; 2024 [citado 12 Mar 2024]. Disponible en: https://www.nicklauschildrens.org/conditions/oral-facial-digital-syndrome [Buscar en Google Scholar]
- Syed S, Sawant PR, Spadigam A, Dhupar A. Oro-facial-digital syndrome type I: a case report with novel features. Autops Case Rep. 2021;20(11):e2021315 [Buscar en Google Scholar]
- Ko Y, Ko J, Ro Y, Kim J. Oral-facial-digital syndrome type 1: A case report and review. Ann Dermatol. 2022;34(2):132-5 [Buscar en Google Scholar]
- Shashikant GA, Lamba G. A case of oro-facial digital syndrome. Pan African Med J. 2022;43(151):34814 [Buscar en Google Scholar]
- Taboada N, Lardoeyt R. Utilidad del método clínico en el diagnóstico de algunos síndromes genéticos. Rev Cubana Med Gen Integr [Internet]. 2018 [citado 12 Nov 2023];34(4):[aprox. 10p]. Disponible en: https://revmgi.sld.cu/index.php/mgi/article/view/539/227 [Buscar en Google Scholar]
- Taboada LN. El consentimiento informado en la práctica asistencial e investigativa de la Genética Clínica. Act Med Centr [Internet]. 2017 [citado 23 Oct 2023];11(3):[aprox. 12p]. Disponible en: https://revactamedicacentro.sld.cu/index.php/amc/article/view/775/1058 [Buscar en Google Scholar]
- Papuc SM, Erbescu A, Glangher A, Streata I, Riza AL, Budisteanu M, et al. Autistic Behavior as Novel Clinical Finding in OFD1 Syndrome. Genes. 2023;14(2):327 [Buscar en Google Scholar]
- Pezzella N, Bove G, Tammaro R, Franco B. OFD1: One gene, several disorders. Am J Med Genet C Semin Med Genet. 2022;190(1):57-71 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129