
Presentaciones de casos
Manifestaciones cutáneas en la enfermedad de la orina con olor a jarabe de arce: reporte de un caso
Cutaneous Manifestation Maple Syrup Urine Disease: Case Report

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-06-05 19:43:30
Aprobado: 2024-06-25 23:59:28
Correspondencia: Gabriel Alejandro Díaz Bernal. Hospital Pediátrico Universitario José Martí Pérez. Sancti Spíritus. gabrieldb@infomed.sld.cu
RESUMEN
Palabras clave: lesiones cutáneas; aminoácidos esenciales; reporte de casos
ABSTRACT
Key words: skin lesions; essential amino acids; case report
INTRODUCCIóN
La enfermedad de la orina con olor a jarabe de arce (EOOJA), también conocida como leucinosis, es una entidad autosómica recesiva producida por un error congénito en el metabolismo de tres aminoácidos esenciales de cadena ramificada (AACR): valina, leucina e isoleucina.(1)
Presenta una frecuencia media en todo el mundo de 1/185.000 recién nacidos, aunque existen grupos étnicos con incidencia tan alta como 1 en 200 nacimientos. Es más frecuente en poblaciones con alta tasa de consanguinidad, como en los Menonitas y en el Oriente Medio.(2) En Cuba, según cifras del Centro Nacional de Genética Médica(3,4) desde 2008 hasta 2022 solo se han reportado 6 casos de la enfermedad, de ellos, dos han sido publicados como casos clínicos en la literatura nacional.(5,6)
Desde el punto de vista clínico, se describen cuatro subtipos fenotípicos solapantes de la enfermedad: clásica o neonatal y las formas intermedia, intermitente y sensible a tiamina, de aparición más tardía.(4,7,8) Las manifestaciones clínicas son olor de los líquidos orgánicos similar al “jarabe de arce o caramelo quemado” (particularmente intenso en el cerumen) y enfermedad fulminante en los primeros días de vida, que inicia con vómitos y letargo y progresa a convulsiones, coma y muerte. Los pacientes con formas más leves de la enfermedad pueden manifestar síntomas solo durante episodios de estrés (por ejemplo: en infecciones o ante cirugías).(9)
A veces pueden aparecer lesiones cutáneas inespecíficas y secundarias al difícil manejo dietético-nutricional. Se manifiestan clínicamente por pápulas eritematosas y descamación blanquecina fina con tendencia a confluir formando placas; predominan en áreas periorificiales y pliegues y son semejantes a las observadas en estados carenciales.(10,11)
En el laboratorio, el diagnóstico se establece a partir de la presencia de niveles elevados de aminoácidos ramificados en plasma (leucina, valina e isoleucina) y de aloisoleucina. En orina, se detectan niveles aumentados de los 2-hidroxiácidos y 2-cetoácidos correspondientes.(4)
Se tiene en cuenta como tratamiento a largo plazo, la restricción de aminoácidos de cadena ramificada, se considera el trasplante hepático como curativo.(9)
Debido a la baja incidencia de esta entidad en Cuba, con menos de una decena de casos reportados en los últimos 15 años, el escaso porciento de supervivencia en la forma neonatal y lo poco descrito sobre la acrodermatitis dismetabólica en estos pacientes, la presente investigación se propone enriquecer la literatura científica nacional e internacional, al aportar un nuevo caso que sirva de base para futuras investigaciones.
PRESENTACIóN DEL CASO
Se presenta el caso de un recién nacido, de sexo masculino de 10 días, con antecedentes pre, peri y posnatales normales, que fue remitido de su área de salud por quejido respiratorio, ganancia insuficiente de peso y pobre succión, además de orinas escasas, polipnea ligera y tiraje intercostal, con la impresión diagnóstica de bronconeumonía.
Al llegar al Servicio de Neonatología ubicado en el Hospital General Universitario Camilo Cienfuegos de la provincia Sancti Spíritus se examinó y se constató recién nacido hipertónico, con dificultad para abrir los puños, rígido a la manipulación y con llanto patológico débil, movimientos involuntarios, además se constató una parada respiratoria, por lo que se interpretó como una convulsión neonatal. Se procedió de manera inmediata a la realización de maniobras o medidas de emergencia (catéter umbilical, dosis de fenobarbital, intubación y ventilación mecánica invasiva).
Se decidió su ingreso en la Unidad de Cuidados Intensivos Neonatales (UCIN) con el diagnóstico de: recién nacido a término con peso adecuado para su edad gestacional más convulsión neonatal.
Complementarios positivos al ingreso:
- Glucemia: 1.9mmo/l.
- Gasometría e ionograma: se interpretó como resultado una acidosis metabólica descompensada con hiperoxia, hipercalcemia e hipercloremia.
- Rayos X de tórax (anterior-posterior) (AP): mostró área cardiotímica dentro de límites normales, condensación inflamatoria hacia base pulmonar derecha con imágenes hiliofugales en ausencia congénita de pericardio en ambos campos pulmonares (ACP).
Durante su evolución presentó acidosis metabólica difícil de corregir, asociada a trastornos hidroelectrolíticos de difícil manejo, así como edema cerebral a las 24 horas del ingreso, con hipertonía y rigidez mantenida. Debido a la persistencia de la acidosis metabólica, el cuadro convulsivo y el edema cerebral, alrededor de las 48 horas de estadía en la UCIN, se planteó la posibilidad de un error innato del metabolismo (12 días de vida) y se tomaron muestras para análisis de laboratorio.
Se recibió desde el Centro de Nacional de Genética Médica un perfil metabólico de ácidos orgánicos con aumento marcado de metabolitos, sugestivo de la enfermedad de la orina con olor a jarabe de arce (EOOJA) (niveles aumentados de ácido 2-hidroxi-valérico, 2-ceto-isovalérico, 2-hidroxi-isocaproico, 2-ceto-isocaproico).
Con el diagnóstico establecido de la EOOJA se impuso el abordaje de fase aguda, dado por la restricción del aporte de aminoácidos de cadena ramificada (AACR), se suspendió la lactancia materna que se sustituyó por fórmulas lácteas especiales y la administración por vía parenteral de glucosa al 10 % en combinación con lipofundin, un preparado destinado a proporcionar energía y ácidos grasos polinsaturados, se garantizó un aporte calórico entre 120 – 130 Cal/kg/día; así como coctel vitamínico (tiamina, riboflavina, piridoxina, cianocobalamina, L-carnitina, ácido fólico). El paciente tuvo una respuesta adecuada a la terapéutica, sin necesidad del uso de métodos de depuración.
A los 34 días de vida, debido a su estadía prolongada en la ventilación artificial mecánica y su estado nutricional, se discutió el caso en colectivo para decidir la realización de traqueostomía y gastrostomía para un mejor manejo dietético y favorecer el destete, porque además presentó pérdida total del reflejo de deglución. Llevó durante su estadía seguimiento estrecho con manejo nutricional intensivo por parte de nutrición pediátrica, consumió por vía enteral (gastrostomía) MSUD-Anamix Infant (0.7kcal/ml-2g de equivalente proteico); un preparado nutricional en polvo para niños entre 0-12 meses de edad, exento de isoleucina, leucina y valina, mezcla de aminoácidos esenciales y no esenciales, hidratos de carbono, lípidos, vitaminas, minerales, elementos traza y fibra, sin lactosa ni gluten.
Se decidió el traslado del paciente al Hospital Pediátrico Universitario José Martí Pérez desde los servicios neonatales a los 84 días de vida, con traqueostomía y gastrostomía, para seguir su atención multidisciplinaria y el entrenamiento familiar para la manipulación del paciente y la elaboración de los preparados nutricionales, se le dio el alta a los 15 días de estadía hospitalaria.
Durante sus primeros 14 meses de vida presentó siete ingresos producto a infecciones respiratorias agudas, con infección de las secreciones por cánula de traqueostomía, pero conservaba un adecuado estado nutricional en relación a su edad. Una vez alcanzados los 16 meses de vida se decidió realizar cierre de la traqueostomía y mantener la gastrostomía para un mejor aporte alimenticio, no obstante, desde el año de edad correspondía realizar el cambio de suplemento nutricional a MSUD-Anamix Junior, comprendido para las edades entre 1-10 años por su mayor densidad energética (1.07kcal/ml-10g de equivalente proteico); pero por las dificultades que enfrenta el país para obtener estos productos, no fue posible su adquisición en esta etapa de la vida del paciente. Por esta situación se mantuvo con MSUD-Anamix Infant, por lo que recibió menos aportes energéticos de los correspondientes.
A los 24 meses de vida, asociado a cuadros diarreicos, presentó lesiones en piel eritematopapulosas que evolucionaron a escamosas, con descamación fina que dejó una mácula hipopigmentada de base, localizadas en región distal de ambos miembros superiores e inferiores, pliegue del cuello, axilas y región perianal, las cuales fueron interpretadas clínicamente como acrodermatitis enteropática, sin lograr cuantificar niveles séricos de zinc por no tener disponibilidad de reactivo. Se comenzó tratamiento con zinc vía oral y cuidados dermatológicos orientados por dicha especialidad. El paciente resolvió parcialmente su estado de la piel. (Fig. 1).
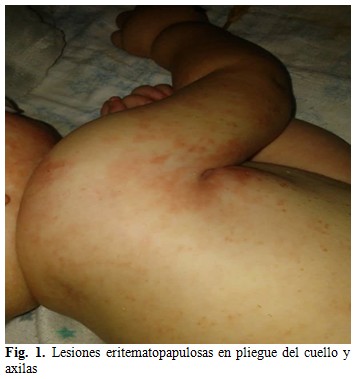
A los 25 meses de edad, un mes después del episodio anterior, fue visto nuevamente en consulta con progresión de las lesiones en la piel, esta vez con tendencia a confluir la formación de placas, con una descamación más extensa, con ligero trasudado. Se realizaron estudios microbiológicos, que fueron negativos. Ante la pobre respuesta al tratamiento anterior impuesto con oligoelementos y la no evidencia de infección por medios de cultivo, se planteó el diagnóstico clínico de acrodermatitis dismetabólica por déficit de aminoácidos, con el inconveniente de no poder cuantificar los aminoácidos de cadena ramificada por no contar con el reactivo. Se realizó un reajuste nutricional de aminoácidos sustentado en su aporte por medio de fuentes naturales, ya que Cuba no cuenta con suplementación de AACR, así como la administración vitamínica y de oligoelementos.
Después de quince días fue llevado a urgencias por un empeoramiento de las lesiones en la piel, que ocupaban ya cerca del 98 % de su superficie corporal, las cuales recordaban un síndrome de piel escaldada y con estado toxinfeccioso, por lo que ingresó en la Unidad de Cuidados Intensivos Pediátricos (UCIP) con antimicrobianos de amplio espectro, fluidoterapia, vitaminoterapia parenteral y soporte nutricional intensivo con alimentación parenteral con dextrosa y lipofundin, MSUD Anamix Infant por vía enteral y suspensión del aporte exógeno de aminoácidos por fuentes naturales, ante el impedimento de realizar determinación sérica de estos. A las 72 horas se acrecentó el deterioro de las lesiones en la piel, que comenzaron a trasudar líquido de aspecto cetrino y con descamación en grandes colgajos, como un gran quemado. Presentó además somnolencia, marcada palidez cutáneo-mucosa, cianosis y frialdad distal, diuresis escasa, hipotensión, intolerancia a los alimentos administrados por gastrostomía; por lo que se planteó el diagnóstico de shock séptico, con posibilidad etiológica de un síndrome de shock tóxico estafilocócico, corroborado por cultivo de las lesiones, que arrojó un staphylococcus aureus meticilin resistente. (Fig. 2).
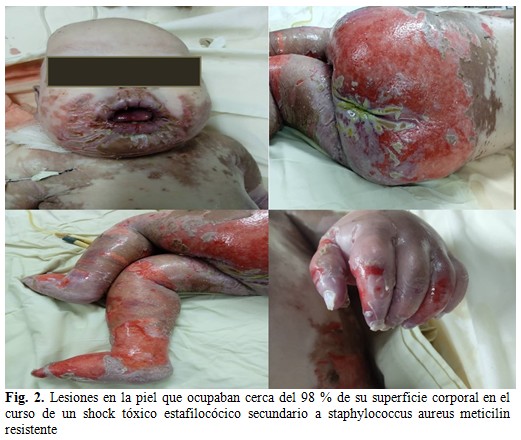
Durante su evolución comenzó con dificultad respiratoria, frialdad distal, hipotermia, anuria, anemia marcada, hipotensión arterial, cianosis generalizada y bradicardia, se realizaron medidas de reanimación cardiopulmonar durante una hora sin lograr actividad eléctrica del corazón por lo que se declaró fallecido.
DISCUSIóN
La enfermedad de la orina con olor a jarabe de arce es un error innato del metabolismo de los aminoácidos de cadena ramificada.
Fue reportada por primera vez en 1954 por Menkes y cols. quienes detectaron cuatro casos en una familia con daño neurológico progresivo de inicio neonatal y con desenlace letal hacia el tercer mes de vida. Los afectados presentaron un olor particular en la orina, semejante al jarabe de arce usado como alimento.(6,8,11) Westall y cols. en 1957, encontraron niveles elevados de aminoácidos de cadena ramificada (BCAA) en sangre y luego Menkes, en 1959, aisló los correspondientes cetoácidos en la orina de estos pacientes y sugirió que el trastorno se debía al bloqueo de la decarboxilación oxidativa de los BCAA, hecho que fue confirmado por Dancis en 1960.(5)
El trastorno aparece por un déficit en la actividad de la deshidrogenasa de los α-cetoácidos de cadena ramificada, complejo enzimático necesario en la segunda reacción del catabolismo de los aminoácidos ramificados: leucina, isoleucina y valina. Este complejo está compuesto por cuatro componentes catalíticos: E1 o descarboxilasa, con dos subunidades E1a y E1ß, que están codificadas por los genes BCKDHA (19q13.1-q13.2) y BCKDHB (6q14.1); E2 o dihidrolipoil transacilasa, codificada por DBT (1p31) y E3 o dihidrolipoil deshidrogenasa, que codifica el gen DLD (7q31-q32).(2,4,8)
La EOOJA sigue un patrón de herencia autosómico recesivo y se produce por variantes patogénicas (homocigóticos o heterocigóticos compuestos) en al menos tres genes: BCKDHA, BCKDHB y DBT,(4) lo que explica sus diferentes fenotipos clínicos.(7,8)
Desde el punto de vista clínico, se describen cuatro subtipos fenotípicos solapantes de la enfermedad: clásica, intermedia, intermitente y sensible a tiamina. La variante clásica se caracteriza por un inicio neonatal: un recién nacido a término, que permanece asintomático durante un período de cinco a diez días, presenta rechazo al alimento y somnolencia y evoluciona al coma inexplicado. El paciente sin tratamiento evoluciona a una falta de regulación vegetativa y en la mayoría de los casos muere durante los primeros meses de vida por crisis metabólicas recurrentes, sin embargo, los pacientes tratados antes de los 10 días pueden llevar una buena calidad de vida, con un coeficiente intelectual normal. Esta variante es la que se describe con mayor frecuencia, se estima que afecta a un 75 % de los pacientes con la EOOJA, como el caso presentado, cuyo diagnóstico se realizó a los 12 días de nacido por un cuadro de convulsión neonatal, edema cerebral y acidosis metabólica de difícil manejo.(3,7) Otras bibliografías(1,5,11) hacen referencia a una quinta forma por deficiencia en la subunidad E3, muy poco frecuente y el cuadro clínico es similar a la forma intermedia, con aumento de los aminoácidos VIL, ácido láctico, ácido pirúvico y α-cetoglutárico. Entre los 2 y 6 meses se produce una importante acidosis láctica, con deterioro neurológico, hipotonía y movimientos anormales.
Los recién nacidos afectados por EOOJA no tienen ningún síntoma ni signo al nacer. El período asintomático puede durar una o dos semanas, en dependencia del grado de deficiencia de la BCKD y no necesariamente de la cantidad de proteínas ingeridas; el catabolismo endógeno proteico causado por el estrés del parto y el ayuno durante las primeras horas de vida pueden ser suficientes para provocar un progresivo aumento de la leucina.(8)
Cuando se le administran proteínas al bebé, normalmente poco tiempo después del nacimiento, como es la lactancia materna, los síntomas tienden a comenzar. Algunos de los primeros síntomas son: falta de apetito, debilidad al mamar, pérdida de peso, llanto agudo, orina que huele a jarabe de arce o azúcar quemada. Los bebés tienen episodios denominados “crisis metabólicas” muchas de ellos no reciben tratamiento adecuado. Algunos de los síntomas de una crisis metabólica son: demasiado sueño, pereza, irritabilidad, vómitos, episodios en los que el tono muscular alterna entre la rigidez y la flaccidez, edema cerebral, convulsiones, acidosis metabólica y coma, que puede terminar en muerte.(5) Gracias a la intervención del equipo multidisciplinario de atención del paciente en la Unidad de Cuidados Intensivos Neonatales (UCIN), con el diagnóstico precoz y tratamiento oportuno, el paciente evolucionó satisfactoriamente en su inicio sin desenlace fatal.
Este cuadro clínico corresponde a una encefalopatía aguda durante los primeros días de la vida neonatal, aunque hay formas más leves que pueden tener su presentación inicial en edades mayores. La combinación de niveles normales de amonio, en ausencia de acidosis metabólica (observada en estados iniciales de la enfermedad) en un neonato en coma, o con signos de encefalopatía, deben aumentar la sospecha de la enfermedad.(8)
El diagnóstico de la EOOJA se realiza a partir de las manifestaciones clínicas típicas, como son los signos neurológicos y el olor a jarabe de arce en la orina y el cerumen. En el laboratorio, el diagnóstico se establece a partir de la presencia de niveles elevados de aminoácidos ramificados en plasma (leucina, valina e isoleucina) y de aloisoleucina. En orina, se detectan niveles aumentados de los 2-hidroxiácidos y 2-cetoácidos correspondientes.(4,5,7,8)
Varios países han introducido la EOOJA dentro de los programas de pesquisa neonatal. En Cuba, el diagnóstico se realiza en pacientes con hallazgos clínicos compatibles con este trastorno. El Laboratorio de Genética Bioquímica del Centro Nacional de Genética Médica implementa dentro de los métodos de laboratorio, la prueba del 2,4-dinitrofenilhidrazina en orina, como un método cualitativo, el análisis de aminoácidos en suero, que ha estado disponible en placa delgada o por cromatografía líquida de alta resolución y el análisis del perfil de ácidos orgánicos en orina por cromatografía gaseosa acoplada a espectrometría de masas (CG-EM).(4)
El objetivo del tratamiento de los pacientes afectados por EOOJA es la rápida normalización de los niveles de BCAA y, en especial, de la leucina por ser el aminoácido ramificado más neurotóxico. Dado que la mayoría de casos son diagnosticados por un cuadro neurológico grave con descompensación metabólica por ser la forma de presentación más común (neonatal clásica), es urgente el tratamiento de la enfermedad en esta fase aguda.(5,7,8)
El manejo terapéutico incluye cuatro premisas fundamentales: suprimir el catabolismo de proteínas endógenas, promover el anabolismo, remover sustancias tóxicas (incluido el amonio) y prevenir las deficiencias de aminoácidos esenciales, mantener los niveles de leucina <200 μmol/l, de valina entre150 – 250 μmol/l y de isoleucina entre 50 – 150 μmol/l.(7) En el presente caso esto fue una limitante, pues no se contó con el reactivo para cuantificar de forma seriada niveles séricos o en orina de aminoácidos, cuestión que en los últimos días de vida repercutió en su manejo personalizado.
En relación a la nutrición, se debe establecer una restricción proteica inicial, entre las 24 a 48 horas de la crisis y después iniciar con un 25 a un 50 % de los requerimientos proteicos para la edad, excepto en el caso de hiperamonemia. Se recomienda el uso de nutrición parenteral con lípidos, dextrosados y aminoácidos, libre de AACR o a través de vía enteral administrar una fórmula libre de AACR, con cantidades calculadas de isoleucina y valina, hasta alcanzar un aporte de 2–3.5 g/kg/día de proteína. Se reintroduce la proteína intacta o aminoácidos, cuando la leucina plasmática se acerque al límite superior del rango de tratamiento.(5,7)
Entre las técnicas de depuración extracorpóreas, la hemodiálisis se considera el manejo de elección, también puede usarse diálisis peritoneal y hemofiltración según experiencia y disponibilidad.(1,5,7,8) Su indicación se da ante niveles de leucina >564 μmol/l (33 mg/dl) y síntomas neurológicos graves sin mejoría clínica tras 24 horas con dieta exenta de AACR.(7,8) El presente caso tuvo una respuesta adecuada a la terapéutica, sin necesidad del uso de métodos de depuración.
La tiamina, se inicia como prueba terapéutica de 1 a 4 semanas a dosis de 50 a 200 mg/día, y se debe mantener en pacientes que responden al tratamiento.
El tratamiento de sostén de la EOOJA, consiste en una dieta que limita la cantidad de AACR, a través de una fórmula láctea especial libre de ellos; control de los niveles de leucina, según rango etáreo y aloisoleucina ausente. En algunos casos es necesario el trasplante hepático; después de un trasplante exitoso los pacientes pueden consumir una dieta sin restricciones.(7,8,9)
Las fórmulas comerciales disponibles se basan en una dieta libre de AACR que brindan un aporte de 2 a 3 g/K/día de equivalente proteico y de 20 a 24 Kcal/oz de aporte calórico.(8) En este paciente se utilizó el MSUD-Anamix Infant (0.7kcal/ml-2g de equivalente proteico); un preparado nutricional en polvo para niños entre 0-12 meses de edad, exento de isoleucina, leucina y valina, mezcla de aminoácidos esenciales y no esenciales, hidratos de carbono, lípidos, vitaminas, minerales, elementos traza y fibra, sin lactosa ni gluten, no obstante, desde el año de edad correspondía realizar el cambio de suplemento nutricional a MSUD-Anamix Junior, comprendido para las edades entre 1-10 años por su mayor densidad energética (1.07kcal/ml-10g de equivalente proteico); pero por las dificultades que enfrenta el país para obtener estos productos de fabricación foránea, no fue posible su adquisición en esta etapa de la vida del paciente. Por esta situación se mantuvo con MSUD-Anamix Infant, por lo que recibió menos aporte energético del correspondiente, lo que repercutió en el desarrollo posterior de estados carenciales.
Como se explicó anteriormente, uno de los pilares del manejo terapéutico de estos pacientes es mantener concentraciones plasmáticas de AACR dentro de un rango de tratamiento objetivo para prevenir las deficiencias de aminoácidos esenciales. En este caso particular, al no contar con la fórmula especial libre de AACR adecuada para la edad, con suplementos de isoleucina que aportaran los 100 mg/día, necesarios, y los reactivos para el frecuente monitoreo sérico o en orina de aminoácidos, trajo consigo un estado de déficit en el que se desarrollaron manifestaciones cutáneas del tipo acrodermatitis dismetabólica, esta entidad es poco tratada en la literatura.(12,13,14,15)
Estas lesiones cutáneas han sido descritas durante el tratamiento de aminoacidopatías y acidemias orgánicas (acidemia metilmalónica, aciduria glutárica y acidemia propiónica) debido a la deficiencia de isoleucina. Se le ha llamado acrodermatitis acidémica o acrodermatitis dismetabólica. Otras causas nutricionales también se han descrito como la deficiencia de biotina y ácidos grasos libres. Por ello, es necesario realizar un seguimiento frecuente de los pacientes, para optimizar su crecimiento y su ingesta dietética y garantizar niveles normales de aminoácidos de cadena ramificada.(12,13,14)
La acrodermatitis dismetabólica (AD) describe erupciones caracterizadas por la tríada clínica de dermatitis acral, diarrea y alopecia, asociada con trastornos metabólicos. La AD es clínicamente idéntica a la acrodermatitis enteropática, sin embargo, resulta por deficiencia de aminoácidos esenciales y ácidos grasos en lugar de zinc.(14,15)
Las lesiones en la piel son inespecíficas y secundarias al difícil manejo dietético-nutricional. Se manifiestan clínicamente por pápulas eritematosas y descamación blanquecina fina con tendencia a confluir formando placas y predominan en áreas periorificiales y pliegues. Las biopsias de piel muestran alteraciones discretas, semejantes a las encontradas en estados carenciales, como los causados por falta de aporte de aminoácidos, ácidos grasos esenciales y déficit de zinc.(10,11)
Este paciente presentó lesiones escamosas periorificiales, que transitaron por varios estadios y empeoraron a pesar de las medidas tomadas. En un primer momento fueron interpretadas clínicamente como acrodermatitis enteropática, asociada a un cuadro diarreico, sin lograr cuantificar niveles séricos de zinc por no tener disponibilidad de reactivo, por lo que se obtuvo una respuesta parcial al tratamiento. Cuando las lesiones progresaron, a pesar de la administración de oligoelementos y en ausencia de infección por medios de cultivo, se planteó el diagnóstico clínico de acrodermatitis dismetabólica por déficit de aminoácidos, con el inconveniente de no poder cuantificar los aminoácidos de cadena ramificada por no contar con los recursos. Aunque se realizó un reajuste nutricional de aminoácidos por medio de fuentes naturales porque en Cuba no se cuenta con suplementación de AACR, entonces las lesiones avanzaron y de manera secundaria fueron colonizadas por estafilococo, lo que provocó un shock tóxico estafilocócico letal a los 2 años de edad. La sobreinfección de estas lesiones ha sido reportada en otros casos similares.(8)
Uaariyapanichkul y cols.(12) presentaron un caso similar, con diagnóstico de EOOJA a los 12 días de vida, y que en sus primeros seis meses tuvo varios episodios de lesiones cutáneas por acrodermatitis dismetabólica y enteropática, que logró recuperar con un manejo estricto del aporte nutricional y la constante detección de concentraciones séricas de aminoácidos.
En la EOOJA, la aparición de lesiones cutáneas probablemente está relacionada con la deficiencia de isoleucina, la cual puede interferir en la síntesis de proteínas esenciales para metabolismo de los queratinocitos. Esto resalta el papel de la deficiencia de isoleucina en la patogénesis de la AD y la importancia de equilibrar cuidadosamente los niveles de AACR durante el tratamiento, ya que la deficiencia puede precipitar la AD.(15)
La EOOJA es un error innato del metabolismo, con una alta tasa de mortalidad en su forma clásica neonatal y que requiere mantener concentraciones plasmáticas de AACR dentro de un rango de tratamiento objetivo, para evitar cuadros de encefalopatía por exceso de aminoácidos neurotóxicos como la leucina o lesiones carenciales del tipo acrodermatitis dismetabólica por déficit de isoleucina.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Roles de autoría:
1. Conceptualización: Gabriel Alejandro Díaz Bernal, Magdalena de la Caridad Guirado Espinosa.
2. Curación de datos: Gabriel Alejandro Díaz Bernal, Magdalena de la Caridad Guirado Espinosa.
3. Análisis formal: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez, Magdalena de la Caridad Guirado Espinosa.
4. Adquisición de fondos: Esta investigación no contó con la adquisición de fondos.
5. Investigación: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez, Magdalena de la Caridad Guirado Espinosa.
6. Metodología: Mányeles Brito Vázquez.
7. Administración del proyecto: Gabriel Alejandro Díaz Bernal.
8. Recursos: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez, Magdalena de la Caridad Guirado Espinosa.
9. Software: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez.
10. Supervisión: Mányeles Brito Vázquez.
11. Validación: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez.
12. Visualización: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez.
13. Redacción del borrador original: Mányeles Brito Vázquez.
14. Redacción, revisión y edición: Gabriel Alejandro Díaz Bernal, Mányeles Brito Vázquez, Magdalena de la Caridad Guirado Espinosa.
REFERENCIAS BIBLIOGRÁFICAS
- Ramírez F, Mato I, Barboza A, Cejas N. Diálisis peritoneal en un neonato con enfermedad de la orina con olor a jarabe de arce. A propósito de un caso. Arch Argent Pediatr [Internet]. 2020 [citado 12 Ene 2023];118(2):[aprox. 8p]. Disponible en: https://www.sap.org.ar/docs/publicaciones/archivosarg/2020/v118n2a22.pdf [Buscar en Google Scholar]
- Barban AV, Araújo W, Fantozzi D, Anderotti G, Aparecida A, Amorín T, et al. Maple syrup urine disease in Brazilian patients: variants and clinical phenotype heterogeneity. Orphanet J Rare Dis. 2020;15(1):309 [Buscar en Google Scholar]
- Camayd I, Nuevas L, Concepción A. Diagnóstico bioquímico de acidurias orgánicas en Cuba: periodo 2008-2013. Act Bioquím Clín Latinoam [Internet]. 2015 [citado 12 Jun 2023];49(2):[aprox. 5p]. Disponible en: https://www.redalyc.org/pdf/535/53541089004.pdf [Buscar en Google Scholar]
- Camayd I, Martínez L, Pacheco B, Concepción A. Diagnóstico de la enfermedad de la orina con olor a jarabe de arce en Cuba en el período 2013-2022 [Internet]. La Habana: Convención Internacional de Salud; 2022 [citado 23 Oct 2023]. Disponible en: https://convencionsalud.sld.cu/index.php/convencionsalud22/2022/paper/download/2617/1357 [Buscar en Google Scholar]
- Busto R, Castellanos ME, Font L, Rodríguez E, Rodríguez B. Enfermedad de la orina con olor a jarabe de arce. Caso único en Cuba. Rev Méd Electrón [i]. 2014 [citado 11 Sep 2023];36(5):[aprox. 10p]. Disponible en: https://www.revmatanzas.sld.cu/revista medica/ano 2014/vol5 2014/tema13.htm [Buscar en Google Scholar]
- Jiménez Y, Esquivel L, Fleites Y, León Y. Necrosis en espejo de ganglios basales en la enfermedad de la orina con olor a jarabe de arce. Presentación de un caso. Medisur [Internet]. 2022 [citado 6 Ene 2024];20(4):[aprox. 5p]. Disponible en: https://medisur.sld.cu/index.php/medisur/article/view/5345 [Buscar en Google Scholar]
- Álvarez AE, Bermejo SM, Stapper SY. Reporte de caso y revisión de literatura: Enfermedad de orina con olor a jarabe de arce. Rev Pediatr [Internet]. 2020 [citado 23 Ago 2023];53(1):[aprox. 5p]. Disponible en: https://revistapediatria.org/rp/article/view/158 [Buscar en Google Scholar]
- Díaz E, Estrada E, Rivera FD. Enfermedad de orina de jarabe de arce: reporte de caso y revisión de la literatura. Rev Colomb Salud Libre [Internet]. 2021 [citado 29 Dic 2023];16(1):[aprox. 15p]. Disponible en: https://revistas.unilibre.edu.co/index.php/rcslibre/article/view/6551 [Buscar en Google Scholar]
- Chávez S, Bravata JC, Sierra M. Errores innatos del metabolismo, una mirada a un tópico poco valorado. Rev Hosp Juárez Mex [Internet]. 2018 [citado 11 Sep 2023];85(3):[aprox. 8p]. Disponible en: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=82474 [Buscar en Google Scholar]
- Becerril G, Ruíz R. Manifestaciones cutáneas de las aminoacidurias. Actas Dermosifiliogr [Internet]. 1998 [citado 12 Mar 2023];89(3):[aprox. 10p]. Disponible en: https://www.actasdermo.org/es-linkresolver-manifestaciones-cutaneas-las-aminoacidurias-13003279 [Buscar en Google Scholar]
- Becerril G, Vela M, Tamayo L, Durán C, Orozco L, Ruíz R, et al. Manifestaciones cutáneas en la enfermedad de la orina con olor a jarabe de arce. Bol Med Hosp Infant Mex [Internet]. 1995 [citado 17 Jun 2023];52(1):[aprox. 5p]. Disponible en: https://www.imbiomed.com.mex/articulo.php?id=28665 [Buscar en Google Scholar]
- Uaariyapanichkul J, Saengpanit P, Damrongphol P, Suphapeetiporn K, Chomtho S. Skin Lesions Associated with Nutritional Management of Maple Syrup Urine Disease. Case Rep Dermatol Med. 2017;39(5):658 [Buscar en Google Scholar]
- Ross B, Kumar M, Srinivasan H, Ekbote AV. Isoleucine Deficiency in a Neonate Treated for Maple Syrup Urine Disease Masquerading as Acrodermatitis Enteropathica. Indian Pediatr. 2016;53(8):738-40 [Buscar en Google Scholar]
- Tabanlioğlu D, Ersoy S, Karaduman A. Acrodermatitis enteropathica-like eruption in metabolic disorders: acrodermatitis dysmetabolica is proposed as a better term. Pediatr Dermatol. 2009;26(2):150-4 [Buscar en Google Scholar]
- Alkhayal FA, Al Haddad S, Bakraa RM, Alqahtani A. Acrodermatitis dysmetabolica secondary to isoleucine deficiency in infant with maple syrup urine disease. Dermatol Reports. 2023;15(4):9750 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129