
Artículos originales
Fenotipo clínico de la fucosidosis en Cuba
Clinical Phenotype of Fucosidosis in Cuba

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-03-14 16:33:57
Aprobado: 2024-07-24 16:20:04
Correspondencia: Víctor Jesús Tamayo Chang. Centro Provincial de Genética Médica. Hospital Pediátrico Universitario Octavio de la Concepción y de la Pedraja. Holguín. vtamayo431@gmail.com
RESUMEN
Objetivo: caracterizar el fenotipo clínico de la fucosidosis en Cuba.
Método: se realizó un estudio de serie de casos, para el que se analizaron las historias clínicas y genéticas de los pacientes con actividad de la enzima α-L-fucosidasa en leucocitos, cuantificada mediante espectrofluorimetría 30 % menor que la actividad de controles sanos. Se describió el comportamiento de variables clínicas relacionadas con el inicio, evolución y conclusión de la enfermedad y se clasificaron los enfermos según los tipos clínicos. Para la descripción de las variables se utilizaron los estadígrafos: frecuencia absoluta, porciento, razón y proporción. Como estadígrafo de tendencia central se empleó la media aritmética y como estadígrafo de dispersión: la desviación estándar.
Resultados: la facies hurleroide, la afectación neurológica y del desarrollo físico, las contracturas articulares y las deformidades esqueléticas fueron las manifestaciones clínicas más frecuentes en los 19 casos diagnosticados. El 84,2 % de los enfermos presentó angioqueratoma y en el 52,7 % existió retraso del desarrollo psicomotor de inicio en la lactancia y neurodegeneración rápida, con pérdida de las habilidades motoras y del lenguaje antes de los cinco años. El 42,1 % de los enfermos falleció entre los 10 y 20 años de edad.
Conclusiones: a pesar de predominar el inicio temprano con retraso precoz del desarrollo psicomotor y la neurodegeneración rápida, el fenotipo clínico de la fucosidosis se comportó como un espectro continuo de severidad, fue la más frecuente una forma clínica de progresión intermedia con fallecimiento durante la segunda década de la vida y presencia de angioqueratoma.
Palabras clave: fucosidosis; retraso del desarrollo; neurodegeneración; angioqueratoma
ABSTRACT
Objective: characterize the clinical phenotype of fucosidosis in Cuba.
Method: a case series study was carried out, for which the clinical and genetic histories of patients with α-L-fucosidase enzyme activity in leukocytes were analyzed, quantified by spectrofluorimetry 30 % lower than the activity of healthy controls. The behavior of clinical variables related to the onset, evolution and conclusion of the disease was described and the patients were classified according to clinical types. To describe the variables, the statisticians were used: absolute frequency, percentage, ratio and proportion. The arithmetic mean was used as a statistician of central tendency and as a statistician of dispersion: the standard deviation.
Results: hurleroid facies, neurological and physical development impairment, joint contractures and skeletal deformities were the most frequent clinical manifestations in the 19 diagnosed cases. 84.2 % of patients presented angiokeratoma, and in 52.7 % there was delay in psychomotor development that began in infancy and rapid neurodegeneration, with loss of motor and language skills before the age of five. 42.1 % of patients died between 10 and 20 years of age.
Conclusions: despite the predominance of early onset with early delay in psychomotor development and rapid neurodegeneration, the clinical phenotype of fucosidosis behaved as a continuous spectrum of severity, was the most frequent a clinical form of intermediate progression with death during the second decade of life and presence of angiokeratoma.
Key words: fucosidosis; delayed psychomotor; neurodegeneration; angiokeratoma
INTRODUCCIóN
La fucosidosis es una enfermedad de almacenamiento lisosomal, causada por la pérdida de actividad de la enzima α-L-fucosidasa, que conlleva a la acumulación de glicoproteínas y glicolípidos fucosilados en varios tejidos y órganos.(1,2) La variedad de hallazgos clínicos que incluye es muy amplio. Además del deterioro neurológico que predomina, se pueden presentar alteraciones dermatológicas, faciales, respiratorias, oculares, cardiacas, disostosis múltiple y visceromegalias, así como afectación del crecimiento y desarrollo.(3,4,5)
La muerte suele ocurrir antes de los 10 años de edad en menos de la mitad de los enfermos y después de los 20 años solo en un 40 % de ellos. Los pacientes con síntomas tempranos tienden a mostrar un deterioro neurológico más rápido, que conduce al fallecimiento en una edad temprana. Se ha documentado que alrededor del 60 % de los pacientes mueren como consecuencia de infecciones respiratorias y las complicaciones neurológicas.(3,4)
De acuerdo a la variabilidad de las manifestaciones clínicas se han descrito dos tipos de la enfermedad:
- Tipo I o rápidamente progresivo: la neurodegeneración conlleva al estado vegetativo y la muerte antes de los 10 años de edad.
- Tipo II: tiene un curso moderado con una progresión más lenta de los síntomas neurológicos y supervivencia hasta la adultez. La mayoría de los pacientes desarrollan angioqueratoma corporis difuso.(2,3,4)
La fucosidosis es una enfermedad autosómico recesiva producida por mutaciones en la estructura del gen de la ɑ-L-fucosidasa-1: (FUCA1), localizado en el locus 1p36.11, que contiene ocho exones que cubren 23 kb y codifica una proteína sin procesar de 461 aminoácidos, 22 de los cuales corresponden al péptido de señal y 439 a la proteína madura.(4) Hasta la fecha se han reportado 38 variantes patogénicas que producen una actividad enzimática baja o ausente.(6,7,8,9)
La correlación fenotipo-genotipo no está bien definida debido a la gran heterogeneidad alélica y la poca repetitividad de las mutaciones en pacientes no emparentados. Esta enfermedad se manifiesta como un espectro clínico continuo, más que poseer dos tipos clínicos mayores, que de hecho estos no son causados por heterogeneidad de locus y se ha propuesto que factores tanto genéticos como no genéticos pueden ser la causa de la gran variabilidad fenotípica.(3,4,6,7)
El tratamiento actual en la gran mayoría de los casos, se limita al manejo de las complicaciones y al apoyo con fisioterapia, sin embargo, los modelos animales generados han esclarecido la neuropatología de la enfermedad y han abierto el camino para posibles terapias. El trasplante de células madre hematopoyéticas se ha aplicado a un pequeño número de pacientes, con estabilización de los síntomas en algunos casos. La terapia de reemplazo enzimático se prueba actualmente en estudios preclínicos, pero aún no se cuenta con ensayos clínicos de terapia génica.(4,10)
Se han reportado aproximadamente 130 enfermos de fucosidosis en todo el mundo, por lo que su prevalencia inferior a 1 por cada 200 000 nacidos vivos la hace incluir dentro de las enfermedades raras(8,11) sin embargo, su frecuencia es alta en regiones del sur de Italia, en poblaciones de origen indo-mexicano de Arizona y Colorado en los Estados Unidos de Norteamérica, en Túnez y en Cuba, donde los afectados se concentran en la provincia Holguín.(3,4,8,12,13,14,15)
Razón por la cual motivó la presente investigación que tiene como objetivo caracterizar el fenotipo clínico de la fucosidosis en Cuba.
MéTODOS
Se realizó un estudio descriptivo, tipo serie de casos, de la totalidad de enfermos de fucosidosis diagnosticados en el período comprendido entre los años 1985 y 2023, en el Centro Provincial de Genética Médica, del Hospital Pediátrico Universitario Octavio de la Concepción y de la Pedraja de la provincia Holguín. La realización de la investigación comenzó a partir de la revisión detallada de las historias clínicas y clínico-genéticas de cada enfermo.
El diagnóstico bioquímico de la enfermedad se basó en la cuantificación de la actividad de la enzima α-L-fucosidasa en leucocitos mediante espectrofluorimetría, realizada en el Departamento de Genética Bioquímica del Centro Nacional de Genética Médica de Cuba. Se asumió como enfermo de fucosidosis cuando la actividad enzimática fue un 30 % menor que la actividad de los controles sanos.(3,15)
Para facilitar el manejo de la información se creó una base de datos primarios en el microprocesador Excel. Para la descripción de las variables se utilizaron los estadígrafos: frecuencia absoluta, porciento, razón y proporción. Como estadígrafo de tendencia central se empleó la media aritmética y como estadígrafo de dispersión: la desviación estándar.
El procesamiento de los datos se realizó con el empleo del paquete estadístico SPSS for Windows. Para el análisis de la relación entre variables se emplearon la medida simétrica V de Cramer y la medida direccional Coeficiente Eta cuadrado: η². Para la evaluación de la concordancia entre las frecuencias esperada y observada se empleó la Prueba de Kappa. Se consideró significación estadística para valores de p ≤ 0,05.
La investigación se llevó a cabo en correspondencia con las regulaciones establecidas en las declaraciones de Helsinki (2013) y Edimburgo (octubre del 2000) para las investigaciones médicas con seres humanos.
RESULTADOS
Características generales de los pacientes con fucosidosis
En el periodo comprendido entre los años 1976 y 2015 nacieron19 niños en la provincia Holguín a los que se les diagnosticó fucosidosis. El 57,9 % de los enfermos (11) pertenecía al sexo femenino y los ocho restantes al sexo masculino. La confirmación bioquímica de la enfermedad se realizó a los 4,9 ± 2,7 años con un rango entre los 1,5 y 10 años de edad. La media de la actividad de α-L- fucosidasa en leucocitos fue del 1,9 ± 2,2% de la actividad de controles sanos, con un rango enzimático entre 0 y 8,3 %. Solo tres enfermos se encontraban vivos al concluir el estudio, por lo que la letalidad de la enfermedad fue de un 84,2 %.
Caracterización fenotípica
- Síntoma inicial
En el 63,2 % de los pacientes la enfermedad se manifestó antes del año de edad. El síntoma de presentación en el 73,7 % de los casos fue el retraso del desarrollo psicomotor (RDPM), pero en el 26,3 % restante existieron manifestaciones clínicas desde el momento del nacimiento ya que dos casos presentaron hipotonía muscular y uno macrocefalia. El inicio de la enfermedad ocurrió a los 0,7 ± 0,5 años de edad, con un rango entre los 0,0 y 1,5 años.
- Síntomas y signos clínicos
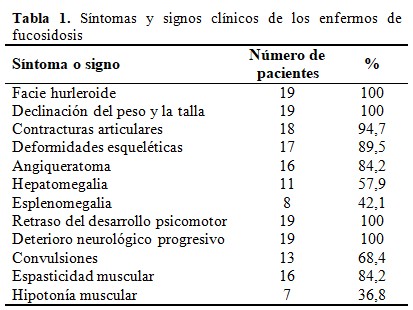
Se muestran a continuación los síntomas y signos clínicos presentes en los enfermos de fucosidosis. (Tabla 1).
- Dismorfia craneofacial
El aspecto facial se caracterizó por la pérdida de la apariencia fina de las cejas, nariz, labios y la cara en su conjunto. La frente solía ser abombada y amplia y se acompañaba de hipertelorismo y estrabismo. La nariz era gruesa, con la punta ancha y redondeada y la raíz ancha y plana. Los labios solían estar engrosados y la boca entreabierta por la presencia de macroglosia. Los dientes presentaban coloración amarillenta y afinamiento de las puntas, malposición y maloclusión y existía hipertrofia gingival.
El conjunto de los rasgos dismórficos daba a la cara un aspecto hurleroide, que fue encontrado en mayor o menor grado en la totalidad de los pacientes y que se comenzaba a hacer evidente a partir de los nueve meses de edad como promedio.
- Desarrollo físico
De forma general los 19 enfermos presentaron afectación del peso y la talla con el transcurso de la enfermedad, sin embargo, solo en el 15,8 % (tres enfermos) existió repercusión en el crecimiento del perímetro cefálico en estadio avanzados de la enfermedad.
- Alteraciones del sistema osteomioarticular
En el 94,7 % de los enfermos las grandes articulaciones fueron afectadas por contracturas en flexión que limitaban el movimiento de los codos, rodillas, muñecas, tobillos, hombros y la articulación coxofemoral. El único paciente que no presentó este signo clínico falleció en etapa temprana de la vida por una enfermedad respiratoria aguda, pero los restantes evolucionaron hacia la cuadriplejia espástica.
Las deformidades esqueléticas incluían fundamentalmente cifoescoliosis, desviación hacia afuera de las manos y pies y defectos vertebrales y costales que llevaban al acortamiento torácico.
- Manifestaciones dermatológicas
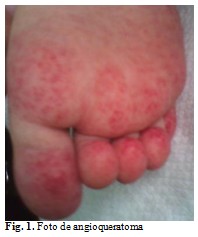
En el 52,6 % de los pacientes se encontró angioqueratoma corporis difuso. En cuatro enfermos la localización del angioqueratoma se circunscribió a la región dorsolumbar y en uno a las palmas de las manos y plantas de los pies. (Fig.1).
- Visceromegalias
La aparición de esplenomegalia siempre se acompañó y fue posterior a la de hepatomegalia.
- Síntomas y signos neurológicos
En el 63,2 % de los casos el inicio del RDPM fue temprano en la etapa de lactantes y en el 36,8 % restante en la etapa de transicionales. El inicio del retraso en la adquisición de las habilidades motoras se evidenció entre los tres y 18 meses de edad y como promedio a los 9,5 meses (0,8 ± 0,5 años). Los cuatro pacientes con inicio más temprano del retraso no lograron el sostén cefálico a los tres meses de edad. Como promedio la sedestación se adquirió a los 10 meses (0,8 ± 0,19 años), la bipedestación a los 12 meses (1,0 ± 0,17 años) y la deambulación a los 21 meses (1,7 ± 0,9 años). Dos pacientes no adquirieron la habilidad de sentarse o mantenerse en la posición de sentados, seis no lograron pararse, y siete no pudieron caminar ni siquiera con apoyo.
La habilidad del lenguaje de al menos palabras bisílabas sueltas se adquirió como promedio a los 17 meses (1,4 ± 1,0 años), pero cinco pacientes no lograron la vocalización. El retraso del desarrollo del lenguaje se hizo evidente entre los ocho y 18 meses de edad con una media de 10 meses (0,9 ± 0,3 años).
En todos de los pacientes existió deterioro neurológico progresivo, con pérdida de la deambulación como promedio a los 5,3 ± 3,5 años de edad en los 12 pacientes que lograron esa habilidad y pérdida del habla a los 5,0 ± 4,1 años de edad en los 14 pacientes que lograron esa habilidad.
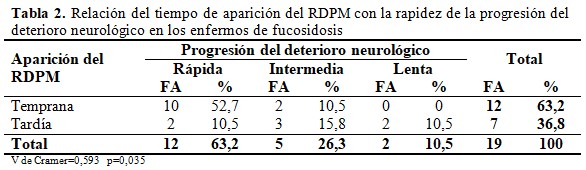
La progresión del deterioro neurológico fue rápida en el 63,2 % de los pacientes, con pérdida completa de las habilidades motoras y del lenguaje antes de los cinco años y lenta en solo el 10,5 % (dos pacientes) con mantenimiento de las habilidades después de los 10 años. En el 26,3 % restante la neurodegeneración fue de progresión intermedia con pérdida de las habilidades entre los cinco y 10 años de edad. Existió una relación estadísticamente significativa entre el inicio del RDPM y la rapidez de la progresión del deterioro neurológico, mientras más temprano fue el retraso en la adquisición de las habilidades psicomotoras más rápida fue la neurodegeneración. (Tabla 2).
La discapacidad intelectual que se presentó en la totalidad de los pacientes fue progresiva y con un grado de severo a profundo, hasta llegar al estado vegetativo.
En el 68,4 % de los pacientes se comprobó clínica y electroencefalográficamente la presencia de convulsiones. El inicio de las crisis ocurrió como promedio a los 4,5 años de edad. En el 57,9 % de los enfermos (11 pacientes) se presentaron crisis tónico-clónicas generalizadas y en dos pacientes crisis de ausencia.
La hipotonía muscular no fue un signo frecuente, solo el 36,8 % de los pacientes la presentaron, además de los dos enfermos en que se evidenció desde el nacimiento, en otros tres se encontró en la etapa de lactantes, sin embargo, la espasticidad muscular se encontró en el 84,2 % de los pacientes, con inicio posterior al año de edad.
- Infecciones a repetición
Las infecciones a repetición se presentaron en la totalidad de los pacientes. Los cuadros bronconeumónicos fueron constantes, aparecieron desde la infancia temprana, se hicieron frecuentes en las etapas tardías de la enfermedad y fueron la causa directa del fallecimiento de los pacientes. Las sepsis orales se presentaron con el transcurso de la enfermedad. Un paciente presentó meningoencefalitis bacteriana a los ocho meses de edad.
- Estado vegetativo y muerte
El estado vegetativo se presentó a los 9,3 ± 4,9 años de edad, con un rango que varió desde los 2,5 a los 21 años. El fallecimiento se produjo a los 11,0 ± 5,4 años de edad, con un rango que varió desde los tres a los 23 años. El 43,8 % de los enfermos falleció antes de los 10 años de edad, el 50 % durante la segunda década de la vida y solo un paciente posterior a los 20 años.
El 41,7 % de los pacientes con inicio de la enfermedad antes del año de edad falleció antes de los 10 años y el 57,1 % de los pacientes con inicio de la enfermedad después del año de edad falleció después de los 10 años. Estadísticamente se encontró una relación de efecto o magnitud fuerte entre la edad de inicio de la enfermedad y la edad de fallecimiento. (Tabla 3).

De acuerdo a la edad de fallecimiento y la aparición de angioqueratoma solo dos casos pudieron incluirse con certeza en el Tipo I de la enfermedad o rápidamente progresivo, por haber fallecido antes de los 10 años de edad sin presentar la manifestación cutánea. Otros cinco enfermos pudieran encuadrarse en ese tipo clínico por su edad de fallecimiento, pero presentaron angioqueratoma. En el Tipo II solo pudieron incluirse dos casos, tanto por su supervivencia después de los 20 años como por la presencia de angioqueratoma, uno de ellos se encontraba vivo al culminar el estudio. El 42,2 % de los enfermos presentó un fenotipo intermedio con fallecimiento entre los 10 y 20 años y con presencia de la lesión dermatológica en siete de ellos. (Tabla 4).
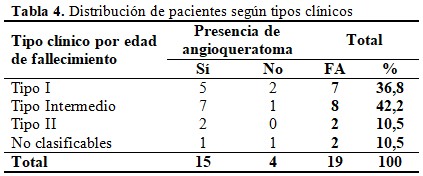
Las dos restantes enfermas no pudieron enmarcarse con certeza en los Tipos I y II de la enfermedad o en la forma clínica intermedia ya que se encontraban vivas en el momento de culminar el estudio, con ocho y 15 años de edad respectivamente, sin angioqueratoma la primera y con la lesión ya establecida la segunda.
DISCUSIóN
Existen pocas publicaciones acerca de la caracterización clínica de la fucosidosis en grupos grandes de pacientes. El estudio más abarcador analizó 77 enfermos diagnosticados en todo el mundo antes del año 1991. Los dos mayores conglomerados hasta esa fecha incluían 20 enfermos procedentes de diferentes regiones del sur de Italia y ocho originarios de la región de nuevo México y Colorado en los Estados Unidos de Norteamérica respectivamente.(3,4) Luego una investigación involucró a 10 pacientes de Túnez diagnosticados entre los años 1987 y 2007.(14)
Las restantes publicaciones solo se han referido a presentaciones de uno o pocos casos,(2, 6,7,8,9,12) por lo que el presente estudio incluye el mayor número de enfermos de fucosidosis con un mismo origen étnico, de nacionalidad y región geográfica de procedencia.
La proporción por sexos observada en la presente casuística: 1 varón por cada 1,375 hembras, no difiere estadísticamente de la proporción esperada de acuerdo al patrón de herencia autosómico recesivo (prueba de concordancia Kappa=0,105; p= 0,516), pero no se corresponde con lo reportado internacionalmente.
Willems, a nivel mundial encontró un exceso de varones (49 varones contra 28 hembras), que mostró una significación estadística en su diferencia con las frecuencias esperadas para cada sexo, a lo que no se le encontró una explicación, aun cuando las frecuencias de presentación del RDPM y el angioqueratoma en cada sexo eran similares si se analizaban como síntomas aislados en la población general.(3) El estudio tunecino a su vez incluía ocho varones y solo dos hembras.(14)
A diferencia de lo observado en el presente estudio, ni la hipotonía muscular ni la macrocefalia se han reportado como signos de inicio de la enfermedad en el momento del nacimiento, pero estos resultados coinciden con lo reportado en relación al RDPM como el síntoma de presentación más frecuente.(2,3,4, 6,7,8.9,12,14)
La ocurrencia de infecciones recurrentes fundamentalmente bronconeumónicas y de convulsiones y la aparición de hepatoesplenomegalia y de una facies hurleroide, se han asociado con el inicio de la enfermedad en una minoría de los casos(3,14) no obstante, en los enfermos holguineros esas manifestaciones fueron posteriores al comienzo del RDPM.
El inicio de la enfermedad en los pacientes del presente estudio fue más temprano que el encontrado en 60 casos a nivel mundial antes de 1991, que fue a los 1,2± 0,8 años,(3) pero es similar al encontrado en Túnez.(14)
Ben Turkia encontró una facies hurleroide en el 90 % de los casos norafricanos, lo que es inferior al hallazgo del presente estudio.(14) El no diagnóstico de los rasgos dismórficos puede estar en relación con el periodo de la enfermedad en que se realiza el examen físico y la minuciosidad con que se buscan, más que con su ausencia definitiva. Las características faciales se deben al acúmulo de sustancias en los tejidos subcutáneos y óseos con el transcurso de la enfermedad.(4)
Existe similitud en los resultados de este estudio con lo reportado en el sumario de 1991, donde se encontró que el 88 % de los pacientes presentaban disostosis múltiple.(3) Stepien reporta esa manifestación en un porciento inferior de casos a nivel mundial (58 %)(4) sin embargo, todos los pacientes tunecinos presentaron alteraciones osteomioarticulares en algún grado.(14) Coincidieron los resultados en la cifoescoliosis como una manifestación frecuente.
La aparición de angioqueratoma se ha asociado con la forma menos severa de la enfermedad o Tipo II, con una supervivencia mayor(3,4) sin embargo, en la casuística holguinera, el 71,4 % de los enfermos que han fallecido antes de los 10 años de edad, presentaron esa manifestación dermatológica, y un caso con desenlace en la segunda década de la vida, no la poseía. Los tipos clínicos de la enfermedad no se pudieron delimitar de forma precisa según la existencia o no de angioqueratoma en Holguín. Estadísticamente se encontró un efecto o magnitud débil de la relación entre la presencia de angioqueratoma y la edad de fallecimiento (Coeficiente de correlación Eta cuadrado η²= 0,234).
Willems, encontró similar contradicción en la relación del angioqueratoma con la edad de fallecimiento y planteó que más bien, el encontrar esa manifestación cutánea dependía de la edad del paciente en el momento del examen. (3) Los pacientes de Túnez con angioqueratoma se correspondían más con la forma clínica intermedia que con el Tipo II de la enfermedad de acuerdo al momento de su fallecimiento y también se planteó que la existencia de la manifestación cutánea era tiempo-dependiente.(14)
La aparición de hepatomegalia en los pacientes holguineros superó el porciento de aparición a nivel mundial (40 %) y en la serie tunecina (20 %). La presencia de esplenomegalia, tuvo un comportamiento similar a lo publicado, en alrededor del 25 % de los afectados.(3,14) Se ha reportado que alrededor del 50 % de los enfermos que presentan hepatomegalia o esplenomegalia poseen ambos agrandamientos viscerales.
El RDPM, la neurodegeneración y la discapacidad intelectual fueron manifestaciones presentes en la totalidad de los pacientes analizados en el presente estudio y esas cifras superan los porcientos de aparición en los pacientes reportados en el sumario mundial de 1991 (88, 88 y 95 % respectivamente), donde los pacientes con ausencia de esas manifestaciones pudieran haberse encontrado en los estadios iniciales de la enfermedad. El porciento de pacientes que lograron sentarse, pararse, caminar y hablar en la casuística cubana es inferior a lo reportado mundialmente, pero la edad en que se adquirieron esas habilidades no difiere.(3)
La presencia de RDPM y neurodegeneración progresiva en todos los pacientes cubanos supera los porcientos de aparición de ambas manifestaciones en Túnez (70 y 60 % respectivamente)(14) sin embargo, ninguno de los 10 afectados del norte de África logró caminar, cifra muy distinta al 63 % de los enfermos holguineros que llegaron a adquirir esa habilidad, pero coinciden los resultados en el 100 % de presentación de la discapacidad intelectual.
Se han reportado enfermos con una afectación moderada del intelecto, e incluso, se describen tres que llegaron a realizar labores manuales en una fábrica(3) sin embargo, la disfunción intelectual en todos los casos del presente estudio fue de grado severo o profundo desde su inicio. Al finalizar la presente investigación, la paciente viva con menor edad se encontraba en estado vegetativo a sus ocho años de nacida.
El mayor porciento de los casos holguineros (63,2 %) tuvo una progresión rápida del deterioro neurológico, a diferencia de lo reportado por Willems, donde solo el 28 % de los enfermos presentaron pérdida completa de las habilidades motoras y del leguaje antes de los cinco años. A su vez, la proporción de casos con deterioro neurológico lento reportada internacionalmente (53 %)(3) supera la de los casos del presente estudio con mantenimiento de las habilidades después de los 10 años, no obstante, mientras más temprana es la aparición del RDPM más rápida es la progresión del deterioro neurológico.
Los porcientos de pacientes reportados en 1991 con epilepsia (38 %) y espasticidad muscular (74 %) fueron inferiores a los encontrados en la casuística cubana, pero la hipotonía tuvo un comportamiento similar.(3)
En todos los casos reportados al igual que en el presente estudio, las infecciones a repetición y fundamentalmente respiratorias tipo bronconeumónicas tienen su comienzo en la infancia temprana y se hacen más frecuentes con la progresión de la enfermedad.(2,3,4,6,7,8,9,12)
A diferencia de lo encontrado en los enfermos holguineros, en los casos de Túnez el estado vegetativo y el fallecimiento son muy próximos. La edad de fallecimiento en seis de esos enfermos fue como promedió a los 42±27 meses, lo cual es más precoz que en Holguín. El 60 % de los pacientes tunecinos que fallecieron antes de los 10 años, supera al 43 % de los pacientes cubanos con igual edad de fallecimiento.(14)
Los pacientes nacidos antes de 1991 presentaron una distribución bimodal de la edad de fallecimiento, con elevaciones antes de los 10 años y después de los 20 años respectivamente y la menor cantidad de defunciones ocurría entre los 10 y 20 años.(3) Esa distribución es distinta a la observada en Holguín, donde el mayor número de pacientes han fallecido en la segunda década de la vida.
Willems encontró que el 82 % de los pacientes con un inicio de la enfermedad antes del año fallecía antes de los 10 años y que el 72 % de los pacientes con inicio de la enfermedad posterior al año de vida fallecía después de los 10 años de edad.(3) Ambas cifras son supriores a las encontradas en el presente estudio, no obstante, se encontró que la edad de fallecimiento guardaba relación con la edad del inicio de la enfermedad.
En Túnez se enmarcó al 60 % de los enfermos dentro del tipo rápidamente progresivo de la enfermedad y al resto dentro del tipo de progresión lenta.(14) Si se incluyeran a los pacientes con el fenotipo intermedio dentro del tipo de progresión lenta, como ocurrió en el estudio tunecino, los porcientos de cada tipo en los pacientes cubanos serían inversos, con casi el 60 % de los enfermos dentro del tipo de progresión lenta.
La delimitación de los Tipos I y II de la enfermedad dentro de los pacientes holguineros se hizo difícil, y el mayor porciento de los enfermos manifestó una forma clínica intermedia, por lo que cabe plantear que la fucosidosis se comporta como un espectro continuo de severidad clínica, con los tipos mayores en sus extremos.
A pesar de predominar un inicio temprano con RDPM en la etapa de la lactancia y una neurodegeneración de progresión rápida, el fenotipo clínico de la fucosidosis en la provincia Holguín se comportó como un espectro continuo de severidad, en el que una forma de evolución intermedia con fallecimiento en la segunda década de la vida y presencia de angioqueratoma fue la más frecuente.
En algunos pacientes no fue posible la realización de estudios imagenológicos del tipo de las resonancias magnéticas nucleares (RMN) que permitieran visualizar con mayor alcance las alteraciones encontradas en el sistema nervioso central (SNC) diagnosticadas en los estudios radiológicos convencionales, por lo que no fueron incluidas en la presente investigación.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Víctor Jesús Tamayo Chang.
2. Curación de datos: Víctor Jesús Tamayo Chang, George Alberto Pérez Benítez.
3. Análisis formal: Víctor Jesús Tamayo Chang, George Alberto Pérez Benítez.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: Víctor Jesús Tamayo Chang, George Alberto Pérez Benítez.
6. Metodología: Víctor Jesús Tamayo Chang, George Alberto Pérez Benítez.
7. Administración del proyecto: Víctor Jesús Tamayo Chang.
8. Recursos: George Alberto Pérez Benítez.
9. Software: George Alberto Pérez Benítez.
10. Supervisión: Víctor Jesús Tamayo Chang.
11. Validación: Víctor Jesús Tamayo Chang.
12. Visualización: George Alberto Pérez Benítez.
13. Redacción del borrador original: Víctor Jesús Tamayo Chang.
14. Redacción – revisión y edición: Víctor Jesús Tamayo Chang, George Alberto Pérez Benítez.
REFERENCIAS BIBLIOGRÁFICAS
- Ferreira CR, Rahman S, Keller M, Zschocke J. An international classification of inherited metabolic disorders (ICIMD). J Inherit Metab Dis. 2021;44(1):164-77 [Buscar en Google Scholar]
- Mao SJ, Zhao J, Shen Z, Zou CC. An unusual presentation of fucosidosis in a Chinese boy: a case report and literature review (childhood fucosidosis). BMC Pediatr. 2022;22(1):403 [Buscar en Google Scholar]
- Willems PJ, Gatti R, Darby JK, Romeo G, Durand P, Dumon JE, et al. Fucosidosis revisited: a review of 77 patients. Am J Med Genet. 1991;38(1):111-31 [Buscar en Google Scholar]
- Stepien KM, Ciara E, Jezela A. Fucosidosis-Clinical Manifestation, Long-Term Outcomes, and Genetic Profile-Review and Case Series. Genes (Basel). 2020;11(11):1383 [Buscar en Google Scholar]
- Kulcsarova K, Baloghova J, Necpal J, Skorvanek M. Skin conditions and movement disorders: hiding in plain sight. Mov Disord Clin Pract. 2022;9(5):566-83 [Buscar en Google Scholar]
- Zhang X, Zhao S, Liu H, Wang X, Wang X, Du N, et al. Identification of a novel homozygous loss-of-function mutation in FUCA1 gene causing severe Fucosidosis: A case report. J Int Med Res. 2021;49(4):3000605211005975 [Buscar en Google Scholar]
- Domin A, Zabek T, Kwiatkowska A, Szmatola T, Deregowska A, Lewinska A, et al. The Identification of a Novel Fucosidosis-Associated FUCA1 Mutation: A Case of a 5-Year-Old Polish Girl with Two Additional Rare Chromosomal Aberrations and Affected DNA Methylation Patterns. Genes (Basel). 2021;12(1):74 [Buscar en Google Scholar]
- Do Rosario MC, Purushothama G, Narayanan DL, Siddiqui S, Girisha KM, Shukla A. Extended analysis of exome sequencing data reveals a novel homozygous deletion of exons 3 and 4 in FUCA1 gene causing Fucosidosis in an Indian family. Clin Dysmorphol. 2023;32(3):112-15 [Buscar en Google Scholar]
- Bhattacherjee A, Desa E, Ahmad K, Jaiswal A, Tyagi S, Dalal A. Genotype first approach & familial segregation analysis help in the elucidation of disease-causing variant for fucosidosis. Indian J Med Res. 2023;157(4):363-6 [Buscar en Google Scholar]
- Kaur A, Dhaliwal AS, Raynes H, Naidich TP, Kaufman DM. Diagnosis and Supportive Management of Fucosidosis: A Case Report. Cureus. 2019;11(11):e6139 [Buscar en Google Scholar]
- Abozaid GM, Kerr K, McKnight A, Al-Omar HA. Criteria to define rare diseases and orphan drugs: a systematic review protocol. BMJ Open. 2022;12(7):e062126 [Buscar en Google Scholar]
- Wang L, Yang M, Hong S, Tang T, Zhuang J, Huang H. Fucosidosis in a Chinese boy: a case report and literature review. J Int Med Res. 2020;48(4):300060520911269 [Buscar en Google Scholar]
- Chkioua L, Amri Y, Chaima S, Fenni F, Boudabous H, Ben H, et al. Fucosidosis in Tunisian patients: mutational analysis and homology-based modeling of FUCA1 enzyme. BMC Med Genomics. 2021;14(1):208 [Buscar en Google Scholar]
- Turkia HB, Tebib N, Azzouz H, Abdelmoula MS, Bouguila J, Sanhaji H, et al. Phenotypic spectrum of fucosidosis in Tunisia. J Inherit Metab Dis. 2008;31(Suppl. 2):S313-6 [Buscar en Google Scholar]
- Tamayo VJ, Llauradó RA, Campos D, Monaga M, Santana EE. Fucosidosis en la provincia Holguín. Causas y frecuencia. Rev Cubana Genet Comunit [Internet]. 2013 [citado 12 Feb 2024];7(2):[aprox. 5p]. Disponible en: https://www.medigraphic.com/pdfs/revcubgencom/cgc-2013/cgc132f.pdf [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129