
Presentaciones de casos
Aplasia cutis congénita. Una serie de tres casos
Congenital Aplasia Cutis. A Series of Three Cases

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2021-03-02 23:48:07
Aprobado: 2021-03-19 18:56:31
Correspondencia: Elsy Roxana Geroy Moya. Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos. roxanagm@hosped.cfg.sld.cu
RESUMEN
Palabras clave: displasia ectodérmica; anomalías congénitas; informes de casos
ABSTRACT
Key words: ectodermal dysplasia; congenital abnormalities; case reports
INTRODUCCIÓN
La aplasia cutis congénita (ACC) fue descrita por primera vez por Cordon en 1767, con dos casos de ACC en extremidades. La ausencia congénita de piel de localización preferente en el cuero cabelludo, es referido por primera vez por Campbell en 1826; el cual describió 2 pacientes con aplasia cutis congénita que afectaba el cuero cabelludo, uno de los cuales murió por hemorragia espontánea del seno sagital superior. En 1833, Billard describió el defecto craneal asociado a este padecimiento.(1,2,3,4)
Es un trastorno poco frecuente y heterogéneo, con una incidencia estimada de 1-3 casos por cada 10.000 nacimientos. La aplasia cutis congénita es típicamente esporádica, pero se han notificado casos autosómicos dominantes y con menos frecuencia autosómicos recesivos, por lo que deben buscarse los antecedentes obstétricos y familiares y por asesoramiento genético buscar miembros de la familia con igual afección. No hay predilección racial o sexual, excepto si está asociada esta última con un síndrome de malformación ligada al cromosoma X.(5,6,7,8,9)
Se han informado tasas de mortalidad que oscilan entre el 20 y el 55 % en asociación con grandes áreas de ACC del cuero cabelludo, a menudo secundaria a hemorragia del seno sagital, complicaciones quirúrgicas, infección o defectos congénitos asociados.(6)
La principal hipótesis fisiopatológica sobre la ACC es que el mecanismo subyacente radica en la ruptura inducida por la tensión de la piel suprayacente que se produce entre las 10 y 15 semanas de gestación, cuando se produce un crecimiento cerebral rápido junto con la dirección y el patrón del cabello lo que explica la alta incidencia de lesiones en el vértice del cráneo. Por otra parte, la ruptura y la formación de bandas amnióticas pueden ser la causa de ACC. Además, se incluyen otras proposiciones: traumatismos intrauterinos, compromiso vascular, infecciones maternas y por el uso de medicamentos.(7,10,11)
El ultrasonido puede predecir la probabilidad de tener una aplasia cutis en el útero, aunque depende del paciente. Se plantea que la aplasia cutis congénita es una causa rara de aumento de la alfafetoproteína prenatal.(8)
Esta afección se caracteriza por la ausencia de epidermis, dermis, y en ocasiones, tejido celular subcutáneo y hueso. Su localización más habitual es el cuero cabelludo, encontrándose un defecto óseo subyacente en el 15-20 % de los casos.(12)
Por lo general puede manifestarse como un defecto único del cuero cabelludo, pero también puede afectar cualquier otra región corporal. En algunos casos la ACC se asocia a otras anomalías físicas o síndromes malformativos.(5)
Se han propuesto varios sistemas de clasificación (Demmel 1975, Sybert 1985, Frieden 1986, Kuster y Traupe 1988, Gorlin y cols. 1990 y Evers y cols. 1995) en un intento por aclarar la herencia, asociaciones, patrones y causas de ACC. Frieden desarrolló el esquema de clasificación más ampliamente aceptado para el ACC con una categorización basada en la ubicación, el patrón, la herencia, las malformaciones asociadas, los teratógenos y los síndromes.(3, 13) (Tabla 1).
Su diagnóstico no es siempre simple. En ocasiones es posible tener una sospecha prenatal mediante el estudio ecográfico en las últimas semanas de gestación, aunque el diagnóstico de esta enfermedad será esencialmente clínico y postnatal. El pronóstico depende de la superficie afectada, del grado de profundidad y la existencia de complicaciones.(6)
La ACC puede aparecer en cualquier parte del cuerpo, sin embargo, en el 84 % de los casos, el defecto se encuentra en el cuero cabelludo y suele ser solitario, presentándose como una lesión oval o circular, solitaria, sin pelo, bien delimitada de 1-2 cm y localizada en vertex. Esta erosión puede ser superficial o presentarse como una ulceración, o bien como una placa cicatricial atrófica o hipertrófica cuando se ha producido su reparación intraútero. La profundidad de la úlcera es variable, puede afectar solamente la epidermis o la dermis superior, o extenderse hasta la dermis profunda, el tejido subcutáneo, y raramente al periostio, el cráneo y la duramadre. Las lesiones ajenas al cuero cabelludo pueden incluir la afectación del tronco y/o las extremidades y suelen ser bilateralmente simétricas, pero también se ha informado una distribución asimétrica. Aunque la mayoría de los defectos son pequeños y superficiales, aproximadamente un 20 % de los casos incluye ausencia de hueso.(6,14)
Actualmente no existe un consenso sobre el manejo terapéutico de la ACC. Su tratamiento va a depender del tamaño, localización, grado de afectación de estructuras subyacentes y riesgo de complicaciones potencialmente letales como la hemorragia del seno sagital, meningitis o alteraciones hidroelectrolíticas. El enfoque multidisciplinario debe ser empleado para obtener resultados óptimos.(3,8)
En estadios iniciales el tratamiento es conservador, sin embargo, en algunos casos hay necesidad de realizar cirugía reconstructiva. Para el tratamiento conservador se recomienda evitar la manipulación innecesaria de la zona afectada y se enfoca a prevenir la infección de las lesiones y mantener una adecuada hidratación con un ambiente óptimo para su eficaz cicatrización.(3, 12)
Se presenta esta serie de casos debido a lo poco frecuente y heterogéneo de este padecimiento y su baja incidencia.
PRESENTACIÓN DE LOS CASOS
Caso 1
Se presenta el caso de un lactante de 9 meses de edad, de sexo masculino, de color de piel negra, hijo de padres no consanguíneos, de procedencia urbana, producto de un embarazo que cursó con anemia por lo que requirió transfusión en el tercer trimestre, parto eutócico, pretérmino, con ruptura prematura de las membranas (RPM) de 72 horas de evolución a las 31,3 semanas y peso de 1550 gramos, Apgar 8-9 por carné de vacunación.
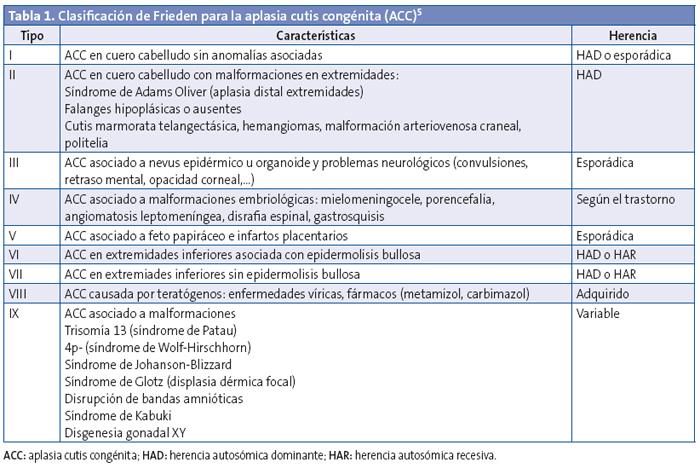
Al nacer se le diagnosticó una aplasia cutis congénita (ACC) con defecto óseo. Se solicitó interconsulta con el Servicio de Neurocirugía, se ingresó en el Servicio de Neonatología y a las 3 horas de nacido comenzó con convulsiones, iniciándose tratamiento con fenobarbital. Requirió ventilación mecánica durante 72 horas, presentó una hemorragia intraventricular Grados 1-2 y una enfermedad de la membrana hialina Grado 2-3, además presentó un íctero fisiológico agravado, por lo cual requirió fototerapia y transfusión en 2 ocasiones. El paciente permaneció hospitalizado 45 días, pendiente de tratamiento neuroquirúrgico para la corrección del defecto óseo y seguimiento en consulta de neurodesarrollo. (Figura 1).
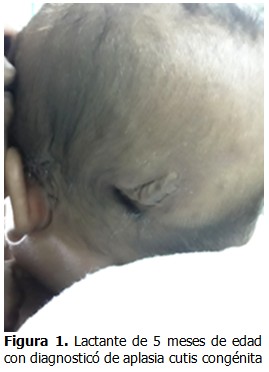
Además, presentó polidactilia izquierda. A los 5 meses fue remitido a consulta de nutrición por pobre ganancia de peso en los 2 últimos meses de vida.
Historia gestacional: primogénito, varón, pretérmino con peso adecuado.
Antecedentes patológicos personales (APP): convulsiones neonatales, hemorragia IV, con presencia de enfermedad de membrana hialina (EMH), íctero agravado, ACC con defecto óseo.
Árbol genealógico: madre con ACC con defecto óseo.
Tías por la línea materna con polidactilia. (Figura 2).
Examen físico
- Evaluación nutricional inicial:
Peso: 5 Kg P/E: 3-10 Percentil.
Talla: 61cm T/E: por debajo del 3er Percentil.
CC: 41cm P/T: por debajo del 3er Percentil.
Edad: 5 meses Pi: 6,4Kg.
2. Datos positivos al examen físico.
- Cráneo: presencia de defecto óseo en región occipital izquierda, con aplasia cutis y pequeño aumento de volumen en región central de la lesión.
- Hipotonía ligera.
- Polidactilia izquierda.
Estudios por imágenes:
Ecografía transfontanelar: mostró discreta dilatación de los ventrículos laterales.
VLD: 5,4 mm. VLI: 5,0 mm. CIH: Ensanchada. Defecto óseo.
Ultrasonido (UTS) abdominal: normal
Ecocardiograma: normal
Tomografía axial computarizada (TAC) de cráneo: mostró atrofia cortical.
Resonancia magnética (RMN) de cráneo: se constató el defecto óseo, presencia de pequeño encefalocele.
Se le plantean entonces los siguientes diagnósticos: lesión estática del sistema nervioso central, de posible etiología prenatal, aplasia cutis con defecto óseo subyacente, encefalocele y polidactilia y una desnutrición P-E.
Se decidió realizar seguimiento multidisciplinario con especialistas de nutrición, neurocirugía, neurología y fisiatría.
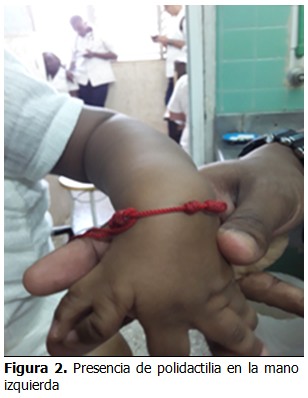
Cuando se logró la recuperación nutricional del paciente se aplicó cirugía correctora del defecto, se realizó la exéresis de la lesión y se colocó un colgajo de piel para el cierre. (Figura 3).
Caso 2
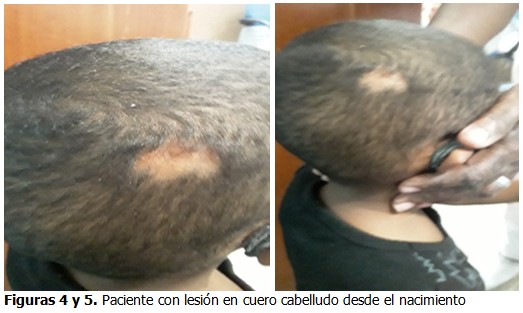
Se presenta el caso de un paciente de 3 años de edad, de sexo masculino, de color de piel negra, hijo de padres no consanguíneos, de procedencia urbana, producto de un embarazo que cursó sin alteraciones, parto distócico por cesárea (iterada anterior), a las 40,5 semanas, peso insuficiente al nacer de 2 700gr. Con lesión en cuero cabelludo desde el nacimiento. Mostró un desarrollo psicomotor normal. Con poca ganancia de peso desde los 9 meses de edad por lo que fue remitido a consulta provincial de nutrición.
Examen físico
1. Evaluación nutricional inicial:
Peso: 11 Kg P/E: 3-10 Percentil.
Talla: 84 cm T/E: por debajo del 3er Percentil.
CC: 47cm P/T: 10-25 Percentiles.
Edad: 3 años Pi: 12,0 Kg.
2. Datos positivos al examen físico:
Cráneo: lesión cicatricial hipertrófica del cuero cabelludo. (Figuras 4 y 5).
Estudio por imágenes:
Ecografíatransfontanelar al nacimiento: normal.
Ecocardiograma: normal.
Ultrasonido (UTS) abdominal: normal.
Historia gestacional:
Varón, con peso insuficiente al nacer.
Antecedentes patológicos personales (APP): no refiere.
Árbol genealógico: nada a señalar.
Se le plantea entonces los siguientes diagnósticos: la presencia de una aplasia cutis congénita Tipo I, con una desnutrición P-E. Con evaluación por dermatología y seguimiento nutricional.
Caso 3
Se presenta el caso de un recién nacido de 15 días, de sexo masculino, de color de piel blanca, hijo de padres no consanguíneos, de procedencia urbana, producto de un embarazo que cursó con infección vaginal en el 1er trimestre, para lo cual llevó tratamiento, parto eutócico a las 39, 2 semanas. Peso al nacer 3 600 gramos. Llegó a consulta de pediatría constatándose al examen físico una lesión cicatricial del cuero cabelludo y soplo sistólico en borde esternal izquierdo bajo, con evaluación con el Servicio de Dermatología, se planteó una aplasia cutis y se le diagnosticó en el Servicio de Cardiología una comunicación interventricular (CIV) pequeña sin repercusión hemodinámica. (Figura 6).

Examen físico
1. Evaluación nutricional inicial
Peso: 3 600 g P/E: 75 Percentil.
Talla: 51cm T/E: 50 Percentil.
CC: 32cm P/T: 50-75 Percentiles.
Edad: 15 días. Pi: 4,0 Kg.
2. Datos positivos al examen físico.
- Cráneo: presencia de una lesión cicatricial en vertex del cuero cabelludo. Fontanelas normotensas.
- Aparato cardiovascular mostró un soplo sistólico en borde esternal izquierdo bajo II-III/VI que se irradia transversalmente.
Estudio por imágenes:
Ecografíatransfontanelar: normal.
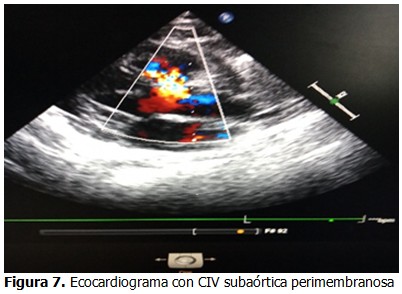
Ecocardiograma: mostró un ligero aumento de las cavidades izquierda, defecto del tabique interventricular de 3 mm en su porción membranosa subaórtica. (Figura 7).
Ultrasonido (UTS) abdominal: normal.
Historia gestacional
Primogénito, varón, con peso adecuado.
Antecedentes patológicos personales (APP): Comunicación interventricular (CIV) sin repercusión hemodinámica, ACC.
Árbol genealógico: Nada a señalar.
Se le plantea entonces los siguientes diagnósticos: una aplasia cutis congénita Tipo I, una CIV subaórtica perimembranosa. Se decidió realizar seguimiento con cardiología.
DISCUSIÓN
La ACC es una característica común de un grupo muy heterogéneo de alteraciones mucho más importantes que la simplemente cutánea, por lo que es necesario realizar una buena historia obstétrica, una meticulosa historia familiar y un cuidadoso examen físico del recién nacido asociado a una batería de pruebas complementarias que permitan descartar malformaciones asociadas.(6)
La ACC solitaria en el cuero cabelludo, es la forma de presentación clínica más frecuente. Puede presentarse como una erosión superficial o ulceración, o bien como una placa cicatricial atrófica o hipertrófica cuando se ha producido su reparación intraútero, como ocurrió en los casos presentados en esta serie.
En un reporte de veintidós casos de ACC publicada por Betancourth-Alvarenga y cols. se constató que las lesiones variaban de 1 cm a 14 cm. Dieciocho casos presentaron lesiones en el cuero cabelludo, 3 en extremidades y uno en tronco.(15)
Su etiopatogenia se desconoce, aunque se ha propuesto que la ACC resulta del desarrollo interrumpido o la degeneración de la piel durante la gestación.(8) Almeida y cols. presentaron una serie de cinco casos que ocurrieron en un período muy breve de tiempo (4 meses) y sin relación aparente. En todos los casos los padres eran mayores de 35 años y no había antecedentes de enfermedad materna o exposición a fármacos durante el embarazo.(5)
En los casos presentados en este estudio solo se identificaron factores genéticos en 1 caso y en los otros dos no se logró identificar los mecanismos por los cuales se produjo la lesión.
La heterogeneidad en los detalles anatómicos y clínicos combinados con las recomendaciones publicadas, a menudo contradictorias sobre el manejo, confunden la toma de decisiones. En aquellos casos con ACC compuesta, el tratamiento quirúrgico precoz con colgajos de rotación del cuero cabelludo se realiza cuando se piensa que es seguro quirúrgica y anestésicamente, mientras que el injerto de hueso primario de los defectos craneales es mejor evitarlos, ya que incluso los grandes normalmente se osifican por completo o dejan solo pequeños huecos óseos de tamaño no crítico. Este enfoque tiene una tasa relativamente baja de complicaciones, minimizando la estancia hospitalaria, proporcionando una función y estética satisfactorias, al reducir el tiempo necesario para asegurar el cierre de la herida.(4)
En la serie de veintidós casos se realizó tratamiento conservador en 9 pacientes y quirúrgico en 13 (8 cierres primarios, 2 plastias, 2 injertos cutáneos y un colgajo).(15)
En el caso 1 de esta serie (ACC con defecto óseo) la cirugía correctora definitiva se realizó cerca de su primer año de vida, mejorando el pronóstico.
La identificación temprana de la lesión, así como la búsqueda de otros síndromes malformativos que pudieran asociarse permitirá que el equipo conformado por neonatólogos, genetistas, dermatólogos, neurocirujanos, cirujanos estéticos y otras especialidades afines, tracen estrategias para evaluar la mejor conducta en cada uno de los casos. Aunque la ACC es típicamente esporádica se recomienda asesoría genética para las familias afectadas por esta enfermedad por la presentación de casos con herencia autosómica dominante o recesiva.
Conflicto de intereses: los autores declaran la no existencia de conflicto de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Elsy Roxana Geroy Moya.
2. Curación de datos: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández.
3. Análisis formal: Elsy Roxana Geroy Moya, Ángel Serafín Camacho Gómez.
4. Adquisición de fondos: Esta investigación no contó con la adquisición de fondos.
5. Investigación: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
6. Metodología: Elsy Roxana Geroy Moya.
7. Administración del proyecto: Elsy Roxana Geroy Moya.
8. Recursos: María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
9. Software: María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
10. Supervisión: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández.
11. Validación: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
12. Visualización: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez
13. Redacción del borrador original: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
14. Redacción revisión y edición: Elsy Roxana Geroy Moya, María Elena Quiñones Hernández, Ángel Serafín Camacho Gómez.
REFERENCIAS BIBLIOGRÁFICAS
- Coronel C, González MD, García S, Bergara M, Guisado MC. Cuatro generaciones de aplasia cutis congénita familiar. Rev Española Pediatr [revista en Internet]. 2017 [citado 23 Mar 2020];73(2):[aprox. 2p]. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=6014460 [Buscar en Google Scholar]
- Elizondo AD, Valdés A. Síndrome de Bart. A propósito de un caso. Rev Argent Dermatol [revista en Internet]. 2017 [citado 2 Feb 2020];98(4):[aprox. 8p]. Disponible en: https://pesquisa.bvsalud.org/portal/resource/pt/biblio-897392 [Buscar en Google Scholar]
- Perry BM, Maughan CB, Crosby MS, Hadenfeld SD. Aplasia cutis congenita type V: a case report and review of the literature. Int J Dermatol. 2017;56(6):118-21 [Buscar en Google Scholar]
- Winston KR, Ketch LL. Aplasia cutis congenita of the scalp, composite type: the criticality and inseparability of neurosurgical and plastic surgical management. Pediatr Neurosurg. 2016;51(3):111-20 [Buscar en Google Scholar]
- Almeida S, Rodríguez F, Coelho S, Bicho MA. Cinco casos de aplasia cutis congénita. Anales Pediatr [revista en Internet]. 2018 [citado 19 Dic 2020];89(5):[aprox. 3p]. Disponible en: https://www.analesdepediatria.org/es-cinco-casos-aplasia-cutis-congenita-articulo-S169540331730485X [Buscar en Google Scholar]
- Clavero N, Orden C, Ochoa L, Berdún E. Aplasia cutis congénita en un recién nacido: tratamiento conservador. Atalaya Médica Turolense [revista en Internet]. 2019 [citado 27 Nov 2020];1(15):[aprox. 4p]. Disponible en: https://atalayamedica.comteruel.org/index.php/revista/article/view/226 [Buscar en Google Scholar]
- Mava Y, Yakubu AM. Aplasia cutis congenita in a Nigerian Child: A case report. J Niger Pediatr. 2017;44(1):32-4 [Buscar en Google Scholar]
- Nyamapfene B, Dube TM, Madondoro K, Musara A, Kalangu K. A Rare Case of Scalp Aplasia Cutis Congenita in a Zimbabwean Child. J Inter Neurosur. 2018;2(2):35 [Buscar en Google Scholar]
- Panda S, Raj R, Mishra P. Not all linear tears are birth injuries: non-syndromic aplasia cutis congenita-a case report. J Med Science. 2016;15(2):19-20 [Buscar en Google Scholar]
- Blionas A, Giakomettis D, Antoniades E, Drosos E, Mitsios A, Plakas S, et al. Aplasia cutis congenita: Two case reports and discussion of the literature. Surg Neurol Int. 2017;8(10):273 [Buscar en Google Scholar]
- Magliah T, Alghamdi F. Aplasia cutis congenita: a case report. Case Reports Dermatol. 2018;10(2):182-6 [Buscar en Google Scholar]
- Higelmo H, Míguez L, Barbato JC, González MT, Reimunde ME, Rodríguez E, Vázquez I. Aplasia cutis congénita con defecto óseo subyacente: evolución favorable tras manejo conservador. J Dermatol. 2018;24(9):1-10 [Buscar en Google Scholar]
- González A, Arriola S, Aguado R, Pérez D, Abreut E. Aplasia cutis congénita, a propósito de un caso. Pediatría Atención Primaria [revista en Internet]. 2020 [citado 12 Feb 2021];22(28):[aprox. 3p]. Disponible en: https://pap.es/articulo/12967/aplasia-cutis-congenita-a-proposito-de-un-caso [Buscar en Google Scholar]
- Shekhar L, Kumble D. Aplasia cutis congenita: A case report. Indian J Case Reports. 2019;(5):50-2 [Buscar en Google Scholar]
- Betancourth JE, Vázquez F, Vargas V, Paredes RM, Ayala J. Manejo quirúrgico de la aplasia cutis congénita. Anales Pediatr [revista en Internet]. 2015 [citado 19 Nov 2020];83(5):[aprox. 5p]. Disponible en: https://www.analesdepediatria.org/ [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129