
Presentaciones de casos
Trombosis espleno-portal no cirrótica, no tumoral y policitemia vera: una asociación poco frecuente
Non-cirrhotic Splenic-portal Thrombosis, non-tumoral Polycitemia Vera: an Infrequent Association
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2018-04-26 22:55:17
Aprobado: 2018-07-03 09:37:24
Correspondencia: Samuel Sánchez Sánchez. Hospital General Universitario Dr. Gustavo Aldereguía Lima. Cienfuegos. samuel92med@gmail.com
RESUMEN
Palabras clave: trombosis de la vena; policitemia vera; enfermedades del bazo
ABSTRACT
Key words: venous thrombosis; polycythemia vera; splenic diseases
INTRODUCCIÓN
La policitemia vera (PV), también conocida como enfermedad de Váquez-Osler se incluye dentro del grupo de las neoplasias mieloproliferativas crónicas, donde no hay presencia del cromosoma Filadelfia (Ph) o su gen de fusión BCR/ABL. Es una neoplasia de células progenitoras hemopoyéticas, con una proliferación celular trilineal, donde predominan las células progenitoras eritroides, con aumento de hematíes circulantes independiente de la eritropoyetina y sin que se reconozca un estímulo fisiológico causal, eritrocitosis primaria.1-6
En su patogenia se invoca el gen JanusCinasa 2 (JAK2). Cursa generalmente con hemoglobina (Hb) y hematocrito (Hto) elevados en forma persistente, cierto grado de leucocitosis, trombocitosis, metaplasia mieloide, tendencia a la fibrosis medular y baja frecuencia de evolución a leucemia aguda. Existe, además, esplenomegalia, hepatomegalia y otros focos de hematopoyesis extramedular.2,5
Aunque es el más frecuente de los trastornos mieloproliferativos crónicos solo se presenta en 2,5 de cada 100 000 personas, afecta a adultos de cualquier edad y se incrementa hasta alcanzar tasas mayores de 10/100 000 personas. Según reportes latinoamericanos, la incidencia en esta área geográfica oscila de 0,5 a 1,5/100,000 personas con una edad media de diagnóstico de 67 años. Puede existir transmisión familiar, pero es poco frecuente, y los casos esporádicos predominan en mujeres.2,5
En los síndromes mieloproliferativos se ha descrito tendencia a la hipercoagulabilidad. Las formas trombóticas de presentación de la PV representan solo entre el 10-15 % del total de casos. Los fenómenos tromboembólicos en miembros inferiores, coronarias, vasos cerebrales y venas suprahepáticas (Síndrome de Budd-Chiari) pueden ser la primera manifestación de la enfermedad, otros sitios son raros y apenas se reportan en la literatura.1-4
El término trombosis venosa portal (TVP) hace referencia a la trombosis que afecta únicamente al tronco portal, se extienda o no a las ramas portales intrahepáticas y el término de trombosis del eje esplenoportal se emplea cuando la trombosis se extiende a la vena esplénica, a la vena mesentérica superior o a la vena mesentérica inferior. La trombosis portal puede ser diagnosticada en el momento agudo (PVT aguda) o puede ocurrir que este evento inicial pase inadvertido y la trombosis portal sea diagnosticada en fase crónica (cavernomatosis portal). La trombosis del eje esplenoportal no asociada a cirrosis hepática o a enfermedad tumoral es la segunda causa de hipertensión portal en el mundo occidental. Hasta en un 60 % de los casos es posible identificar un trastorno protrombótico sistémico subyacente como factor etiológico. Los factores locales son los causantes de un tercio de los casos, y no frecuente la coexistencia de varias entidades. Por eso, en estos pacientes es de vital importancia el diagnóstico etiológico.1-4
La baja incidencia de la policitemia vera y la forma inusual de mostrarse como una trombosis del sistema espleno-portal motivó a la presentación de este caso diagnosticado en el Hospital General Universitario Dr. Gustavo Aldereguía de Cienfuegos.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente de 70 años de edad, de color de piel blanca, de procedencia rural, jubilada, ex fumadora, sin otro hábito tóxico. La paciente no refirió alergias medicamentosas, tampoco cirugías, transfusiones o traumatismos. Se encontraron antecedentes familiares de enfermedades cardiovasculares, y una hermana fallecida hacía poco tiempo de cáncer de páncreas de histología no precisada. Se hallaron antecedentes personales de hipertensión arterial de 15 años de evolución con tratamiento regular con amlodipino (10mg) 1 tableta al día, diabetes mellitus tipo 2 de 14 años de evolución, insulinodependiente de 20 unidades en la mañana y 10 en la noche. Además hacía 9 meses había presentado un infarto agudo de miocardio.
Esta paciente acudió al cuerpo de guardia del Servicio de Medicina Interna por presentar dolor abdominal de 15 días de evolución, de moderada intensidad, de comienzo súbito, persistente, que apenas había cambiado, localizado en epigastrio y con irradiación a hipocondrio izquierdo, que no cedía con analgésicos ni antiácidos y por el cual había acudido en múltiples ocasiones a los servicios de salud sin solucionarse su estado. Se acompañaba de náuseas, disminución del apetito, cefalea y trastornos de la visión. Al examen físico el abdomen resultaba doloroso en hipocondrios y epigastrio, además se palpaba una esplenomegalia discreta. Se decidió realizar su ingreso como una esplenomegalia para estudio.
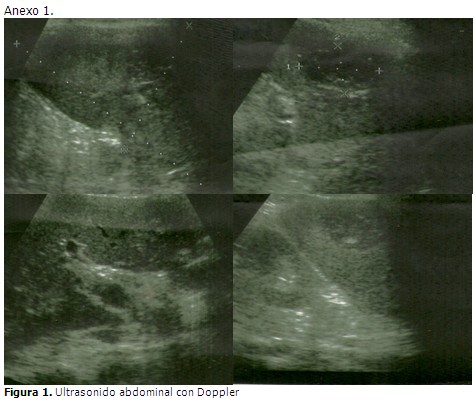
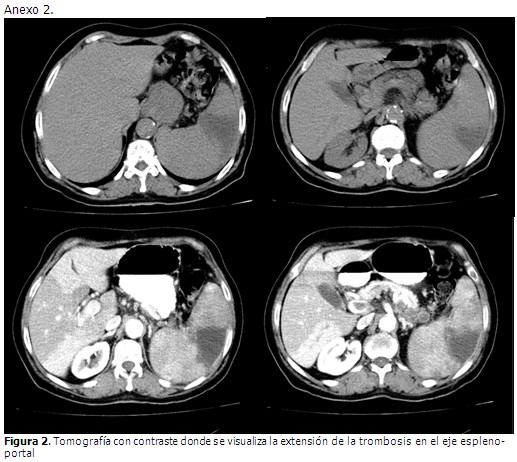
En ultrasonido abdominal realizado se visualizó una imagen compleja en el bazo además de un aumento de tamaño moderado. Posteriormente se realizó un nuevo ultrasonido abdominal, esta vez colegiado, en el que se informó en el hígado, un marcado aumento de la ecogenicidad hacia las paredes de las estructuras vasculares del interior del órgano, a expensas sobre todo de la porta, la cual mostró extensas áreas de trombosis que se extendían hacia el eje espleno-portal; además un aumento del tamaño del bazo de ligero a moderado con imagen ecolúcida en su región central de características no bien precisadas. Dos días más tarde se le realizó una tomografía por contraste que informó hepatomegalia con área hipodensa extensa que respetaba las estructuras vasculares arteriales, con forma irregular que concordaba con el diagnóstico de trombosis venosa espleno-portal, además en el bazo se visualizaba esplenomegalia con área hipodensa irregular en cuña que se extendía de la periferia al centro. Ante la clínica de la paciente y los hallazgos ecográficos y tomográficos se concluyó como una trombosis espleno-portal.
En los estudios iniciales de analítica sanguínea existió un aumento de las transaminasas y de la LDH por encima de los valores de referencia, que con posterioridad mostraron reducción hasta valores normales. No hubo otra manifestación clínica o de laboratorio de insuficiencia hepática.
En la búsqueda de la probable etiología de la trombosis durante el ingreso en sala de se realizó a la paciente: perfil renal, gasometría arterial, coagulograma completo, conteo de Addis, proteinuria de 24 horas, antígeno de superficie para el VHB, anticuerpos contra el VHC, rayos x de tórax y ultrasonido ginecológico con resultados dentro de parámetros normales, ninguno de estos estudios permitió justificar causa paraneoplásica, o séptica de la trombosis.
Desde el comienzo llamó la atención que entre los resultados de analítica sanguínea del día existió una hemoglobina (Hb) de 17,7 g/l y hematocrito (Hto) de 0,57, la paciente refería además que las hemoglobinas anteriores le habían resultado de hasta 18,0 g/l. Retrospectivamente el infarto agudo de miocardio de hacía 9 meses pudo haber sido una manifestación trombótica del proceso al que en este momento se asistía.
Se imponía el estudio de la poliglobulia como causa probable de la trombosis. Se le indicó lámina periférica que informó una leucocitosis moderada con neutrofilia, plaquetas cerca del límite superior. El medulograma mostró hiperplasia de la serie megacariopoyética y concluía como un síndrome mieloprolliferativo crónico. Mientras la biopsia de médula ósea concluía como una hiperplasia de las tres series, la megacariocítica además tenía dismorfia y predominio de formas grandes con tendencia a la agrupación, estudio compatible con proceso mieloproliferativo crónico.
En nuestro medio no se dispone de dosificación de eritropoyetina (EPO). El estudio de la determinación de la mutación JAK2V617F realizada en el Instituto Nacional de Hematología resultó positivo.
Con los elementos anteriores la paciente cumplía con los tres criterios mayores de la Organización Mundial de la Salud del 2016 para la policitemia vera, diagnóstico que se confirmó posteriormente.
Durante su estadía la paciente llevó tratamiento profiláctico de la trombosis con fraxiheparina a dosis terapéuticas y doble antiagregación. Se difirió el uso de heparina sódica y con posterioridad de anticoagulantes orales. Se le practicó flebotomía para la poliglobulia. Tuvo una evolución clínica satisfactoria. En estos momentos se mantiene en seguimiento por la especialidad de hematología.
DISCUSIÓN
Cuando la trombosis del tronco portal se extiende a la vena esplénica, a la vena mesentérica superior o a la vena mesentérica inferior se conoce como trombosis del eje espleno-portal. Puede ser diagnosticada en el momento agudo (PVT aguda) o puede ocurrir que este evento inicial pase inadvertido y la trombosis portal sea diagnosticada en fase crónica (cavernomatosis portal). Es posible afirmar que esta paciente tuvo el diagnóstico en etapa subaguda.7-9
El ultrasonido Doppler es el estudio complementario de elección por su alta sensibilidad y ausencia de efectos secundarios. La realización de la tomografía por contraste y/o la angio-resonancia magnética, son indispensables para establecer la extensión e identificar el momento evolutivo de la trombosis. También permiten identificar la presencia de alteraciones asociadas a la TVP y valorar la presencia de circulación colateral con mayor precisión que la ecografía. En el caso en cuestión se empleó la ultrasonografía y la tomografía por contraste.7-9
El tratamiento de la trombosis incluye la anticoagulación, que debe iniciarse lo más tempranamente posible y debe realizarse con heparina, debido a la rapidez de acción y debe ser mantenida durante al menos de 2-4 semanas, para luego ser sustituida por anticoagulantes orales (con un objetivo de INR 2-3). La anticoagulación debe mantenerse un mínimo de 6 meses, incluso un año. La anticoagulación permanente se recomienda en los casos en los que se identifica un trastorno protrombótico, existan antecedentes personales o familiares de trombosis venosa profunda o historia previa de dolor abdominal que sugiera ser isquémico. Estas pautas internacionales se emplearon en el manejo de la paciente del estudio.7-10
La trombosis portal se encuentra aproximadamente en el 1 % de las necropsias. En la mayoría de las ocasiones esta trombosis se relaciona con cirrosis o neoplasias hepáticas y en tan solo un tercio de los casos es atribuible a un origen no cirrótico y no tumoral. La trombosis del eje esplenoportal no asociada a cirrosis o neoplasias cumple los criterios de enfermedad rara de la Organización Mundial de la Salud (OMS), ya que tiene una prevalencia inferior a 5 por cada 10,000 habitantes.7
Si se realiza un estudio exhaustivo, hasta en un 70 % de los pacientes se logra identificar un factor etiológico causante de la TVP. De estos, hasta en un 60 % de los casos se identifican factores trombogénicos sistémicos, y hasta en un 30 a un 40 % se identifican factores locales predisponentes. Entre el 17-53 % se asocian a un síndrome mieloproliferativo primario, de los cuales el más frecuente es la policitemia vera. Sin embargo en más del 15 % de los pacientes coexisten factores etiológicos múltiples, lo que apoya la teoría que considera la TVP como una enfermedad multifactorial. Debido a la posible y frecuente asociación entre varios factores trombofílicos sistémicos y locales, ante todo paciente con TVP siempre debe realizarse un estudio minucioso de todos los factores etiológicos conocidos. A pesar de todo esto, hasta en un 30 % de los pacientes la causa no llega a identificarse.7-10
Las manifestaciones generales de la enfermedad incluyen facies pletórica debida a eritrosis, así como otras manifestaciones derivadas de la hiperviscosidad, cefalea, vértigo, manifestaciones de ataques transitorios de isquemia, quemosis conjuntival, prurito acuagénico por activación de basófilos por efecto de la mutación JAK2 V617F y que suele aparecer hasta en un 40 % de los pacientes. La eritromelalgia es un síndrome curioso de etiología desconocida que cursa con eritema, ardor y dolor de las extremidades, en especial de las inferiores, la cefalea ocular y la quemosis conjuntival se consideran una manifestación de este síndrome, posible isquemia digital, tendencia a la formación de equimosis, epistaxis, enfermedad acidopéptica o hemorragia del tubo digestivo a consecuencia de la estasis vascular o la trombocitosis, fatiga, hipertensión sistólica, esplenomegalia palpable hasta en un 70 % de los casos, hipertensión pulmonar, síntomas derivados del hipermetabolismo en especial la intolerancia al calor, derivados de la hiperuricemia como son la gota y la litiasis renal. La organomegalia puede producir malestar mecánico notorio, hipertensión porta y caquexia progresiva. La paciente refirió cefaleas frecuentes, astenia y al examen físico del ingreso presentaba una esplenomegalia moderada que fue corroborada luego por ecografía abdominal, en exámenes paraclínicos posteriores presentó hiperuricemia.1-4
Solo de un 10 a 15 % se diagnostica en el contexto de un evento trombótico arterial o venoso, aunque son las complicaciones más frecuentes y principal causa de muerte en los pacientes con PV. Un tercio se produce antes del diagnóstico y pasa inadvertida por el personal médico, aquí es posible incluir el infarto del miocardio que había presentado la paciente con anterioridad. Dos tercios de las trombosis son arteriales (cerebrales, cardíacas, mesentéricas, etc.) y dentro de las trombosis venosas, las más frecuentes son las TVP de miembros inferiores. El 25 % involucra vasos cerebrales y abdominales, generalmente las supra-hepáticas que causan un síndrome de Budd-Chiari. La trombosis venosa intraabdominal es en particular común en mujeres jóvenes y puede ser catastrófica si ocurre una obstrucción súbita y completa de la vena hepática.1,2
La eritrocitosis puede encontrarse como un hallazgo en laboratorio de rutina o en el estudio de síntomas generales o trombóticos, como ocurrió en este caso. Sin embargo a menos que la concentración de hemoglobina sea ≥20 g/100 mL (hematocrito ≥60 %), no es posible distinguir una eritrocitosis verdadera de trastornos que producen contracción del volumen plasmático. Una característica peculiar de la PV, a diferencia de las demás causas de eritrocitosis verdadera, es que la expansión del volumen plasmático puede ocultar el incremento de la masa eritrocítica, en consecuencia, resulta necesaria la cuantificación tanto de la masa eritrocítica como del volumen plasmático para determinar la presencia de eritrocitosis absoluta y diferenciarla de la relativa, que deriva de una reducción aislada del volumen plasmático (también conocido como eritrocitosis por estrés, espuria o síndrome de Gaisböck ).1-4
Los estudios para la PV incluyen: hemograma completo con índices hematimétricos y frotis de SP, perfil férrico, LDH, ácido úrico, gases arteriales y saturación O2, niveles de EPO sérica; si son elevados es poco probable el diagnóstico de PV y si son bajos son altamente sugestivos de PV (sensibilidad y especificidad del 90-95 %) y excluyen eritrocitosis secundaria (ES). La medición de tamaño de hígado y bazo por imágenes, eritrocitos marcados con Cr y albúmina con I, permiten evaluar el volumen total y masa eritrocitaria en casos dudosos. La dosificación de EPO y la evaluación de masa total no se pudieron realizar por no contar con los reactivos necesarios.1-7
Las técnicas para reconocer la mutación de JAK2 en presencia de saturación normal de oxígeno arterial es otra forma diagnóstica para identificar eritrocitosis cuando no se puede conocer la masa eritrocítica ni practicar las cuantificaciones del volumen plasmático. El descubrimiento de una mutación somática puntual (JAK2V617F) en el gen Januscinasa 2 (JAK2) de la familia de las tirosinas, realizado en los primeros años del actual siglo no solo aportó nuevos elementos en la fisiopatología de la PV, sino que también ha modificado los criterios diagnósticos de esta enfermedad. La Organización Mundial de la Salud (OMS) en el 2001 y 2008, realizó una revisión y propuso nuevos criterios diagnósticos con el objetivo de crear parámetros clínicos, morfológicos y moleculares para incrementar la sensibilidad y la especificidad diagnóstica, así como disponer de algoritmos aplicables a la práctica clínica habitual. Una nueva modificación en estos criterios ocurrió en el 2016.1,2,5
La presencia de la mutación JAK2V617F en la PV varía según diferentes estudios realizados, se informa una frecuencia del 65 - 97 % de acuerdo con la sensibilidad del método empleado, el tipo de muestra a utilizar y el uso de leucocitos totales o granulocitos solamente. La positividad de la mutación JAK2V617F se ha asociado a una mayor edad de diagnóstico de la enfermedad y se señala una media de 59 años. La biopsia de médula ósea no solo se utiliza para confirmar diagnóstico de PV sino que es útil evaluar el grado de fibrosis con fines pronósticos. Hallazgos típicos son la hipercelularidad con hiperplasia de los tres sistemas y con megacariocitos pleomórficos, gigantes, polilobulados y agrupados.2,5
En el tratamiento de la PV se recomienda la corrección de los factores de riesgo cardiovasculares (cese del hábito de fumar, control del peso, de presión arterial y de la glucemia, uso de estatinas en caso de dislipidemias, un plan de ejercicios físicos acordes a la edad y función cardiovascular). Son objetivos del tratamiento la prevención de las complicaciones trombóticas y hemorrágicas, encaminadas a evitar su repetición. El segundo objetivo se encamina a minimizar el riesgo de transformación a leucemia aguda y/o fibrosis, así como el control de síntomas.1-5
La prevención de la trombosis se realiza en primer lugar por flebotomías, cuyo objetivo fundamental es mantener un Hto< 45 %, se ha demostrado que con esto se reducen las muertes por eventos cardiovasculares y trombosis mayores. Generalmente se comienza con 250 a 500ml día con reposición de volumen con solución fisiológica con una frecuencia que depende de la situación clínica del paciente.10-12
Todos ellos deben recibir dosis bajas de AAS (80-100 mg/día) para prevención y tratamiento de trombosis arteriales. En casos de alto riesgo de trombosis, algunos autores recomiendan duplicar la dosis de aspirina (100 mg cada 12 horas). El resultado del uso de otros antiagregantes como las tienopiridinas (clopidogrel, ticlopidina, prasugrel) no es aconsejado, excepto en alergia o intolerancia a la aspirina; no hay estudios que confirmen la seguridad y eficacia de su uso. En caso de efectos adversos gastrointestinales por la AAS, se demostró que es mejor su uso combinado con inhibidor de bomba de protones que cambiar por clopidogrel. En caso de trombosis arterial y/o venosa bajo antiagregación, se debe asociar tratamiento citorreductor.10-12
Para la citorreducción se emplea la hidroxiurea, droga de primera línea que posee un amplio rango de dosis-respuesta, efectos colaterales leves y bajo riesgo mutagénico. La dosis de inicio aconsejada es de 15 a 20 mg/kg/día y se debe regular la dosis de mantenimiento según el hemograma (0,5-1 g/d). Se debe controlar cada 2 semanas en los primeros 2 meses, luego en forma mensual y cada 3 meses cuando se alcanza la dosis estable. Los efectos tóxicos adversos están relacionados a mielosupresión y úlceras orales y en miembros inferiores. El interferón es otra opción, puede utilizarse el interferón-α convencional o pegilado (peg-IFN-α-2a ó 2b) que tiene menos efectos secundarios, es mejor tolerado y de aplicación semanal. El peg-IFN induce remisión hematológica completa en el 54 a 76 % de los casos, mejora los recuentos de plaquetas y leucocitos, mejora la esplenomegalia (77 %), disminuye el prurito refractario y logra respuesta molecular en un 54 %, sin detección del JAK2V617F en el 14 %. Los efectos colaterales más frecuentes son: enfermedades autoinmunes, depresión, síndrome gripal y enfermedades oculares.10-12
El anagrelide tiene uso limitado al reducir las plaquetas. No tiene efecto antiproliferativo, no es leucemogénico, no produce efectos displásicos. Tiene efectos colaterales por su acción inhibidora de la fosfodiesterasa: inotropismo positivo y vasodilatación con palpitaciones, cefaleas, edemas por aumento de la permeabilidad vascular, disnea e insuficiencia cardíaca congestiva, también produce fatiga, nauseas, diarreas, mareos, inestabilidad y algunos casos de alucinaciones. Es necesario el monitoreo de la función cardiaca y está contraindicado en pacientes con enfermedad miocárdica con disminución de la fracción de eyección del VI menor del 50 %.1,10-12
ANEXOS
REFERENCIAS BIBLIOGRÁFICAS
- Spivak J. Polycythemia Vera and Other Myeloproliferative Neoplasm. En: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J. Harrison´s Principles of Internal Medicine. 19 ed. NewYork: McGraw-Hill; 2015: p. 672-8 [Buscar en Google Scholar]
- Tefferi A. Policitemia Vera, trombocitopenia esencial y mielofibrosis primaria. En: Goldman L, Schafer A. Goldman-Cecil Medicine. 25 ed. Philadelphia: Elsevier Saunders; 2016: p. 244-30 [Buscar en Google Scholar]
- Kaushansky K. Hematopoyesis y factores de crecimientos. En: Goldman L, Schafer A. Goldman-Cecil Medicine. 25 ed. Philadelphia: Elsevier Saunders; 2016: p. 237-44 [Buscar en Google Scholar]
- Armitage J, Bierman P. Aproximación al paciente con adenopatias y esplenomegalia. En: Goldman L, Schafer A. Goldman-Cecil Medicine. 25 ed. Philadelphia: Elsevier Saunders; 2016: p. 567-89 [Buscar en Google Scholar]
- Benz EJ. Disorders of Hemoglobin. En: Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J. Harrison´s Principles of Internal Medicine. 19 ed. New York: McGraw-Hill; 2015: p. 178-83 [Buscar en Google Scholar]
- Gisslinger H, Gotic M, Holowiecki J, Penka M, Thiele J, Kvasnicka HM, et al. Anagrelide compared with hydroxyurea in WHO-classified essential thrombocythemia: the ANAHYDRET Study, a randomized controlled trial. Blood. 2013;121(10):1720-8 [Buscar en Google Scholar]
- Pardanani A. Systemic mastocytosis in adults: 2015 update on diagnosis, risk stratification, and management. Am J Hematol. 2015;90(3):250-62 [Buscar en Google Scholar]
- Llop E, Seijo S. Treatment of non-cirrhotic, non-tumoral portal vein trombosis. Gastrohep. 2016;39(6):403-10 [Buscar en Google Scholar]
- Zandrino F, Musante F, Gallesio I, Benzi L. Assessment of patients with acute mesenteric ischaemia: multislice computed tomography signs and clinical performance in a group of patients with surgical correlation. Minerva Gastroenterol Dietol. 2006;52(3):317-25 [Buscar en Google Scholar]
- Orr DW, Harrison PM, Devlin J, Karani JB, Kane PA, Heaton ND, et al. Chronic mesenteric venous thrombosis: evaluation and determinants of survival during long-term follow-up. Clin Gastroenterol Hepatol. 2007;5(1):80-6 [Buscar en Google Scholar]
- Squizzato A, Romualdi E, Middeldorp S. Antiplatelet drugs for polycythaemia vera and essential thrombocythaemia [CD-ROM].Cochrane Database Syst Rev; 2008;16(2): p. CD006503 [Buscar en Google Scholar]
- Harrison C, Kiladjian JJ, Al-Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787-98 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129