
Artículos de revisión
La epistasia en genes modificadores: factor clave para diferenciar subtipos clínicos en los trastornos del neurodesarrollo
Epistasis in Modifying Genes: Key Factor to Differentiate Clinical Subtypes in Neurodevelopmental Disorders
 ,1
,1
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-04-04 05:53:34
Aprobado: 2024-07-08 23:56:00
Correspondencia: José Ignacio Lao Villadóniga. Instituto de Formación Continuada de la Universidad de Barcelona. Sociedad Española de Genética Clínica y Dismorfología. Sociedad Española de Medicina Antienvejecimiento y Longevidad. Director Médico en Genomic Genetics International. Barcelona. España. doctorlao@genomicgenetics.com
RESUMEN
Palabras clave: trastornos del neurodesarrollo; trastorno del espectro autista; genética; genómica; epistasis genética; polimorfismo genético; genes modificadores
ABSTRACT
Key words: neurodevelopmental disorders; autism spectrum disorder; genetics; genomics; genetic epistasis; genetic polymorphism; modifying genes
INTRODUCCIóN
Los trastornos del neurodesarrollo en su conjunto son muy complejos y suelen tener su origen en alteraciones que afectan el desarrollo cerebral. Suelen manifestarse a través dificultades cognitivas de diverso grado, una gran variedad de problemas de conducta y alteraciones motoras de intensidad muy variable de un caso a otro. En muchos de los trastornos del neurodesarrollo las manifestaciones están presentes de por vida y tienen un impacto severo en el funcionamiento normal del cerebro, lo que conlleva a menudo grandes problemas emocionales y físicos, además de económicos, no solo para el individuo, sino también para la familia y la sociedad, de ahí la relevancia de poder diferenciarlos y de esta forma, actuar de forma temprana para atenuar sus repercusiones.
Existen evidencias suficientes para considerar que, en los trastornos complejos del neurodesarrollo, tales como los trastornos del espectro autista (TEA), la discapacidad mental, la parálisis cerebral y el trastorno por déficit de atención con o sin hiperactividad (TDA/H), existe una interacción entre factores genéticos y ambientales donde la epigenética desempeña un papel tan o incluso más crítico, que la propia genética del individuo.(1)
Solo si se consideran estas variables en su conjunto, sin olvidar su constante interacción dinámica, que abarca desde las fases preconcepcionales hasta todas las etapas del embarazo y la postnatal inmediata, se tendrá la posibilidad real de lograr su control, e incluso, garantizar su prevención. Precisamente, como primer paso para la prevención, el desafío está en poder identificar a cada persona especialmente vulnerable para actuar de manera preventiva de forma oportuna y así evitar el impacto negativo del efecto de los factores modificables para los que resulta especialmente vulnerable, según el potencial de base de su genoma.
DESARROLLO
Gracias a los avances en las técnicas de secuenciación masiva, se han identificado muchos genes asociados directamente con los trastornos complejos del neurodesarrollo, y aunque todavía hay mucho por aprender en esta área debido a su heterogeneidad y complejidad, en la actualidad se pueden utilizar muchos de los genes identificados para explicar las diferencias que se pueden encontrar en la mayoría de los casos, no obstante, se puede ampliar el análisis hacia otros genes que según sus versiones (polimorfismos genéticos) actuarían como modificadores al imprimir diferencias de expresión clínica en cada caso, independientemente de que sea catalogado como sindrómico o no, por lo que ayuda a definir estrategias personalizadas de atención que ayuden a mejorar el pronóstico final.
En este sentido, se pueden considerar como genes modificadores, a aquellos genes con determinadas variaciones, consideradas polimorfismos, que al actuar de forma aislada no causan efectos fenotípicos graves en su portador, al no afectar directamente su capacidad de supervivencia, sino que, interactúan con otros genes y con factores no genéticos, son capaces de modular la expresión fenotípica final al marcar la variabilidad observada dentro de un grupo de individuos con un mismo diagnóstico clínico. (Fig. 1).
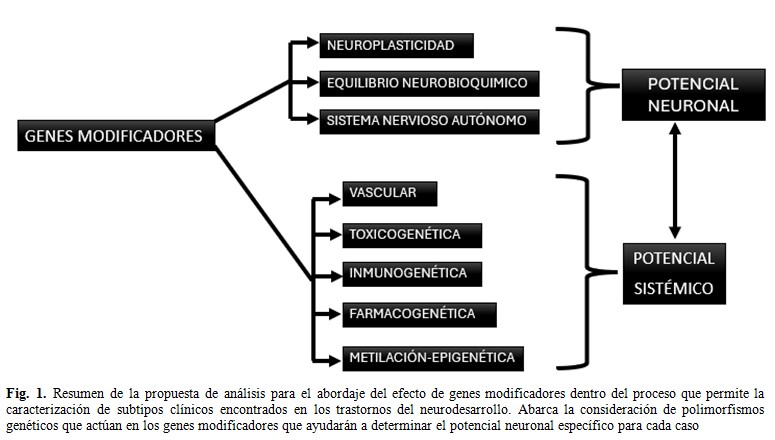
Su importancia radica en que su conocimiento y consideración permitirá ganar precisión tanto a la hora de abordar el diagnóstico, como para el diseño de estrategias de prevención y/o tratamientos con mayor seguridad y eficacia.
Es esencialmente en estos genes modificadores, que se encuentran las diferencias interindividuales que ayudará a definir los endofenotipos que forman los subtipos clínicos que se encuentran dentro de cada diagnóstico clínico.
Una de las herramientas que, en esta línea, ya suele aplicarse es el NeuroProgram, que es reconocida como una herramienta auxiliar que permite llegar a definir subtipos, tanto dentro del TEA, como en el abordaje de cualquier trastorno del neurodesarrollo.(2)
Por otra parte, para valorar el potencial sistémico y su capacidad de interacción con el potencial neuronal, se consideran otros polimorfismos en genes modificadores pertenecientes al potencial de salud vascular, la toxicogenética, la inmunogenética, la farmacogenética y la capacidad de metilación del ADN, que es el mecanismo más estable dentro de los mecanismos moleculares de la epigenética.
Con relación a genes modificadores cuya expresión tiene implicaciones directas en el sistema nervioso, se destacan los siguientes:
1. Polimorfismo en el gen de la apolipoproteína E (APOE) y su implicación en la salud neurológica
Diversos estudios llevados a cabo en modelos celulares y moleculares aportan una serie de datos relevantes que, además, están en consonancia con los hallazgos anatomopatológicos y la propia observación clínica. Estos estudios apuntan a ciertas moléculas cuya relevancia está en que participan tanto en la formación, como en la remodelación y el ajuste funcional de las sinapsis cerebrales. Entre ellas se encuentra la proteína secretada reelina y la vía de señalización que esta media a través de los receptores de lipoproteínas como la Apolipoproteína E (ApoE).(3)
El gen de ApoE, que se representa como APOE, está localizado en el brazo largo del cromosoma 19 y se muestra polimórfico en humanos: presencia de los alelos APOE ε2, APOE ε3 y APOE ε4.
De acuerdo con los resultados de diversos estudios, se ha podido saber que, quienes porten el alelo ε4 (el 27 % de los individuos de la población caucásica) suelen tener niveles elevados de colesterol total y LDL-colesterol y muestran, además, menor respuesta al tratamiento con estatinas y una reactividad exagerada a los cambios de la dieta.
Por otro lado, la presencia de la variante APOE ε4 es considerada como el principal factor de riesgo genético para la enfermedad de Alzheimer de inicio tardío (>65 años). Ha sido ampliamente documentado que el alelo ε4 de la ApoE confiere riesgo para el desarrollo de Alzheimer de manera "dosis-dependiente", es decir, mientras una copia de dicho alelo supone un factor de riesgo de 3.6, la presencia de dos copias aumenta el riesgo hasta 18.(4)
Con relación a los trastornos del neurodesarrollo y especialmente con el TEA, existen múltiples evidencias que convierten al APOE en uno de los genes modificadores que no se deben dejar de analizar si se quiere ganar precisión, tanto en su delineación diagnóstica precisa, como para encaminar toda estrategia de soporte destinada a su control. En este sentido, hay investigaciones que han revelado, que en los portadores del APOE ε4 pueden verse alteradas las conexiones en el cerebro, incluso, en edades tempranas. En adultos este proceso es demostrable mucho antes de que el deterioro cognitivo sea evidente.(5)
La ApoE es muy abundante en el líquido cefalorraquídeo, por lo que se demuestra que cada una de sus variantes presenta una habilidad diferente en el proceso de detoxificación, en especial el de detoxificar el mercurio. Los portadores del APOE ε4 son los más afectados al exponerse a los metales pesados, por lo que es uno de los principales factores que determinarían un peor pronóstico, sobre todo a nivel del deterioro cognitivo.(6)
Con relación a la versión APOE ε2, si bien se ha señalado que es un factor de neuroprotección en adultos, hay estudios que han mostrado que los niños cuya alteración neurológica es más grave al momento del nacimiento tienen mayor tendencia a ser portadores de ese alelo.
Otro estudio, analiza la contribución de APOE en el trastorno de espectro autista (TEA), este estudio mostró que los portadores de la combinación APOE ε2/ ε2 tenían un mayor riesgo frente al daño neuronal durante el período prenatal y la primera infancia. Confirmado este hecho, otros investigadores han informado la asociación del estado de portador de APOE ε2 con resultados adversos tempranos del desarrollo neurológico después de la cirugía cardíaca, independientemente de los factores operatorios y del paciente. Esto implica que los bebés y niños menores de cinco años portadores de APOE ε2, que experimentan alguna lesión que afecte al SNC, tendrían más probabilidades de desarrollar complicaciones neurológicas, sin embargo, esta situación mejora en la edad adulta, por lo que representaría un mejor pronóstico a largo plazo (efecto neuroprotector) que aquellos portadores de APOE ε4 ya que, en estos casos, el riesgo sigue siendo invariablemente alto independientemente de la edad.(7)
En la etapa adulta, APOE ε2 tendría un efecto neuroprotector, como suele reportarse por diversos investigadores; lo que, incluso en la etapa más avanzada, permite atribuirle un papel promotor de la longevidad y de protección contra la demencia. También se ha asociado el APOE ε4 con la menor capacidad de recuperación tras un traumatismo craneal por lo que sus portadores son más vulnerables al daño neuronal postraumático. Parece que el APOE ε4 es capaz de alterar o interrumpir las conexiones de la red neuronal al interferir con la actividad normal de algunos receptores moleculares.
Respecto al riesgo de otros trastornos del desarrollo, la frecuencia de APOE ε4 está particularmente elevada en niños con parálisis cerebral (PC), especialmente con tetraplejia/triplejia, un hallazgo que ha sido reportado independiente del peso al nacer. El estado de portador del alelo e4 también se ha asociado con una mayor gravedad de la PC y con una mayor tendencia a la microcefalia. También la variante APOE ε2 ha sido vinculada al mayor riesgo para la PC entre otros problemas del desarrollo del sistema nervioso, aunque este efecto solo tendría lugar en la vida prenatal.(8)
Si bien el efecto negativo de la variante APOE ε4 sería tanto pre como postnatal, el de APOE ε2 sería más bien perjudicial únicamente en las fases prenatales y neonatales tempranas, por lo que resulta beneficioso a partir del nacimiento.
2. Genes modificadores CNTNAP2 y MET y su papel en la neuroplasticidad
La disección cuidadosa de las contribuciones genéticas a aspectos discretos de la estructura y función del cerebro (los llamados endofenotipos), es una forma de comenzar a desentrañar las bases de las variaciones de la cognición y el comportamiento entre humanos.
Se puede definir la neuroplasticidad como la potencialidad del sistema nervioso de modificarse para formar nuevas redes neuronales en respuesta a cada nueva información, a la estimulación sensorial, al desarrollo y para readaptarse ante la disfunción o el daño. Suele asociarse al aprendizaje que tiene lugar en la infancia, pero sus definiciones van más allá y tienen un recorrido mucho más largo cuyos inicios están en las primeras fases prenatales.
Existen diversos componentes bioquímicos y fisiológicos detrás de un proceso de neuroplasticidad y esto lleva a diferentes reacciones biomoleculares químicas, genómicas y proteómicas que requieren de acciones intra y extra neuronales para generar una respuesta neuronal. Aquí se destaca la plasticidad sináptica que depende de la expresión genética de proteínas de remodelación y la modulación de factores neurotróficos. Entre estos factores se destaca una proteína de la familia de las neurexinas conocida como CNTNAP2 (proteína asociada a contactina 2) y la proteína codificada por el gen MET(9) ambas vinculadas con las señales bioquímicas necesarias para la formación de los circuitos neuronales.
Este es un gen cuyo producto (la proteína CNTNAP2) resulta esencial para garantizar la integridad estructural y funcional del cerebro, porque actúa desde fases muy tempranas del desarrollo del sistema nervioso. Los portadores de estas variantes en CNTNAP2, presentan una falta de conexión entre el lóbulo frontal y otras áreas del cerebro que son importantes para el lenguaje.(10)
Uno de los polimorfismos genéticos que ha sido ampliamente analizado, el conocido como rs7794745, se ha asociado con una mayor susceptibilidad a los trastornos del espectro autista y muchos otros trastornos del neurodesarrollo debido a una capacidad alterada para promover la conexión entre las dos neuronas y la producción de una sinapsis.
Estudios basados en escáner del cerebro en niños que portaban la variante de riesgo, se mostró que el lóbulo frontal estaba mayoritariamente conectado entre sí, en lugar de con las otras zonas del cerebro, como era de esperar de forma habitual. Además, esta región estaba conectada solo localmente en una red bilateral difusa en quienes portan estas combinaciones de riesgo, mientras que en aquellos con el alelo sin riesgo la corteza prefrontal medial transmitía información a regiones posteriores a través de una red en el lado izquierdo. Esta conexión anteroposterior funcional lateralizada izquierda es la que precisamente involucra las regiones del cerebro que controlan el procesamiento del lenguaje. De ahí que quienes porten las combinaciones de riesgo mostrarían trastornos del desarrollo del lenguaje como manifestación principal. En los portadores de las variantes de riesgo es posible que la falta de transferencia eficiente de información a estas regiones desde las áreas frontales pueda contribuir a una mayor probabilidad de que se vean afectados por el TEA u otros trastornos relacionados. También se ha descrito un subtipo caracterizado por un trastorno lingüístico semejante al trastorno específico del lenguaje (TEL) pero sin manifestaciones TEA, no obstante, aunque de forma excepcional, hay casos con estas combinaciones donde puede aparecer un desarrollo precoz de la capacidad de lectura (hiperlexia).(11)
Dentro de la evolución de las manifestaciones, curiosamente se menciona que en alrededor del 20 % de los casos, tras un período de desarrollo normal hasta un promedio de 2 a 5 años, aparece una regresión (cognitiva) que afecta, en primer lugar, al lenguaje, que provoca una pérdida paulatina de las habilidades lingüísticas adquiridas hasta ese momento. En aquellos casos que logran adquirir una competencia lingüística aparentemente normal en lo concerniente a la fonología, la morfología o la sintaxis, se sigue manteniendo un déficit característico en lo que se refiere al componente pragmático del lenguaje.(12)
Por otra parte, hay casos en los que también se describe afectación de la conducta social. Básicamente, se ha comprobado que su interferencia afecta la migración de las neuronas, al interrumpir el crecimiento neuronal en la corteza cerebral y provocar un efecto similar en el cerebelo, por lo que, además, puede llevar a afectación a nivel de la coordinación motora.
Entre todos los genes que contribuyen a la formación correcta del circuito cerebral hay otro particularmente interesante, el gen MET. El gen MET codifica un miembro de la familia de proteínas del receptor tirosina quinasa. Cuando este receptor se une al factor de crecimiento de hepatocitos, se dimeriza y desempeña un papel en la supervivencia celular, la embriogénesis y la migración e invasión celular. En el cerebro humano, este gen se expresa altamente en las porciones temporal, occipital y medial de los lóbulos parietales. Además de su papel relevante en el crecimiento y la maduración neocortical y cerebelosa, su acción resulta clave para garantizar la reparación gastrointestinal y la competencia inmunológica, funciones que curiosamente, también se han reportado como muy afectadas en niños con TEA. En este sentido, es un elemento clave para tener en cuenta en aquellos casos donde las manifestaciones intestinales aparecen como comorbilidad casi constante del cuadro neurocognitivo, si además coexiste con otras variantes genéticas, esta vez a nivel de un perfil inmunogenético favorecedor, con las que entraría en sinergia para reforzar la susceptibilidad a desarrollar complicaciones de salud intestinal.(13)
Específicamente, se ha demostrado que la variante del promotor MET rs1858830, alelo "C", está vinculado a una transcripción génica reducida. Esta variación genética (polimorfismo genético) se reportó originalmente en el año 2006, fue en esta fecha cuando se demostró que su presencia confiere un aumento multiplicado por 2 en el riesgo para el TEA. El estudio estuvo basado en el análisis de ~ 700 familias.(14)
Dada su expresión tanto a nivel del cerebro, como en el intestino, es una de las mejores evidencias que permiten afirmar que hay subtipos de TEA donde se pueden encontrar manifestaciones a estos niveles no como comorbilidad, sino más bien, como parte del cuadro clínico distintivo de este subtipo.(15)
3. Genes modificadores con influencia en el mantenimiento del equilibrio neurobioquímico
- La vía dopaminérgica
La dopamina es concebida como uno de los neurotransmisores catecolaminérgicos más importantes del sistema nervioso central (SNC) se le relaciona con la regulación de diversas funciones motoras, neuroendocrinas, motivacionales, efectivas, así como con el consumo de drogas altamente adictivas como la cocaína, las anfetaminas y otros psicoestimulantes.
En las regiones prefrontales del cerebro determina la eficacia de importantes funciones mentales, como la atención y la memoria, por esta razón ha sido la vía más estudiada a la hora de abordar las causas neurobioquímicas de los trastornos del neurodesarrollo que afectan el potencial cognitivo, la esfera afectiva y el comportamiento social.(16)
En el análisis de esta vía, se deben considerar varios polimorfismos genéticos que, al actuar en sinergia, tienen la capacidad de afectarla tanto por hiperactividad de la propia vía como, por el contrario, la depresión de la vía o hipodopaminergia.
En primer lugar, se debe destacar al polimorfismo genético que afecta el funcionamiento del receptor alfa-adrenérgico 2A (ADRA2A). ADRA2A actúa en la regulación de la liberación de neurotransmisores desde los nervios del sistema simpático y las neuronas adrenérgicas. Existe un polimorfismo de relevancia en el gen que lo codifica, el rs1800544. En un estudio poblacional amplio se detectó que los portadores del genotipo G/G tenían puntuaciones significativamente más altas en las escalas para la evaluación de la depresión y las puntuaciones significativamente más bajas respecto a moral y el orden en comparación con los portadores del genotipo C/C. Las niñas con genotipos C/C y C/G puntuaron más alto que los niños con los mismos genotipos en una evaluación del comportamiento extrovertido. Los chicos con genotipos G/G, sin embargo, tuvieron una mayor puntuación que las niñas con genotipos GG. El alelo G se asocia con la mayor probabilidad de respuesta positiva al tratamiento con metilfenidato. Por otra parte, el genotipo GG se suele asociar con mayor tendencia al déficit de atención.(17)
En la ruta dopaminérgica, el papel de la enzima catecol-o-metiltransferasa (COMT) ha centrado diversas investigaciones teniendo en cuenta el papel de sus variantes en la etiología de varios trastornos psiquiátricos incluidos procesos psicóticos, afectivos y trastornos de ansiedad. El enzima COMT se encarga de la degradación de la dopamina en la región frontotemporal del cerebro. De esta forma, al actuar junto a otros genes en esta vía, la enzima COMT se encarga de mantener el equilibrio en los niveles de la dopamina sináptica. De dichos niveles dependerá la eficiencia de la transmisión sináptica en neuronas dopaminérgicas que a su vez determinará la capacidad para resolver problemas mentales complejos o para afrontar situaciones novedosas y conflictivas.(18)
Además, para cubrir lo más posible la evaluación del potencial individual de la vía dopaminérgica, también se deben analizar polimorfismos con implicación funcional en genes que codifican receptores dopaminérgicos como el DRD2(19) y el gen de la proteína transportadora de la dopamina SLC6A3.(20)
- La vía serotoninérgica
La serotonina (5-HT) es un actor fundamental en el desarrollo del cerebro y los trastornos neuropsiquiátricos. En este sentido, se ha demostrado que los factores que causen desequilibrios a nivel de los niveles de serotonina durante el embarazo son capaces de inducir alteraciones en los circuitos talamocorticales y límbicos prefrontales y en el eje hipotálamo-pituitario-adrenocortical del feto en desarrollo. Como consecuencia, en la vida postnatal, estas alteraciones se han asociado con riesgos de desarrollar trastorno por déficit de atención con hiperactividad, trastornos del espectro autista, depresión y/o ansiedad. Esta es una de las bases que apunta hacia el nexo entre la vía serotoninérgica y los trastornos del neurodesarrollo, con especial interés en el trastorno de espectro autista.
Estudios como el de: Aaron E, Montgomery A, Ren X, Guter S, Anderson G, Carneiro AMD, titulado: Whole Blood Serotonin Levels and Platelet 5-HT2A Binding in Autism Spectrum Disorder, han demostrado que algunos niños con TEA carecen del pico de producción de serotonina visto en niños con desarrollo normal (neurotípicos). Aunque periféricamente (plaquetas) se encuentren altos los niveles de serotonina, se ha mostrado una reducción significativa en su capacidad de unión con los receptores 5HT2 en la corteza cerebral de individuos con TEA y sus familiares de primer grado, mostrándose que su nivel dentro del SNC era inversamente proporcional a sus niveles periféricos.
Por tanto, resulta de interés evaluar los polimorfismos genéticos que afectan la capacidad para mantener el equilibrio de las vías dopaminérgica y serotoninérgica como parte de la estrategia integral que permitirá delinear de forma más precisa cada uno de los casos que se tengan que diagnosticar y tratar con estas manifestaciones.
4. Genes modificadores de acción indirecta sobre el sistema nervioso como modificadores del fenotipo clínico final
Se debe considerar la repercusión sobre el fenotipo clínico final que pueden tener otros genes modificadores, al actuar de forma indirecta sobre el sistema nervioso. Aquí destacan tanto, los genes de diferentes mediadores inmunitarios que determinarían el perfil de las interleucinas vinculadas con linfocitos Th17 y la vía antinflamatoria T-reguladora, como los sistemas de detoxificación enzimática de fase I y fase II a nivel hepático.
La disfunción inmunitaria como clave de diferenciación clínica
Las evidencias resultan suficientes para demostrar que el sistema inmunológico y el sistema nervioso mantienen una interacción casi constante. Además de lo mencionado con anterioridad con relación al gen MET, ampliamente documentado en muchos casos con TEA, varios investigadores han demostrado que existe una relación muy evidente entre la disfunción inmunitaria y los trastornos del neurodesarrollo como el TEA y el TDA/H.
Se consideran perfiles de riesgo en varias de las citocinas incluidas en la vía Th17, cuya hiperfunción se ha asociado a procesos inflamatorios crónicos y autoinmunes, en este programa de evaluación se destacan a los que determinan la actividad de la IL-6, IL-1, TNF-α y IL23R, y además, los que afectan a interleucinas con la capacidad de inhibir o limitar el funcionamiento de las células T reguladoras (Treg), se destaca tanto a la IL-10 como a los que afectan el funcionamiento del receptor de la vitamina D (VDR). Existen estudios como el de: Li DJ, Tsai CS, Hsiao RC, Chen YL, Yen CF, titulado: Associations between Allergic and Autoimmune Diseases with Autism Spectrum Disorder and Attention-Deficit/Hyperactivity Disorder within Families: A Population-Based Cohort Study, basados en metaanálisis que muestran menor concentración de citoquinas antiinflamatorias IL-10 en casos TEA.
La baja capacidad de detoxificación y su implicación en la diferenciación clínica
Una de las observaciones que más se repiten en los casos con TEA y ante los trastornos del neurodesarrollo en general, es la alta tendencia a que se acumulen tóxicos en su organismo. Algo que se constata al hacer el análisis de sus concentraciones como parte de las analíticas que se les indican como parte de su evaluación clínica. Esto pone de relieve contemplar la posibilidad de que estos casos tengan un compromiso funcional importante en sus sistemas enzimáticos de detoxificación.
La oxidación es un proceso natural que está ocurriendo constantemente en todo el organismo pues se necesita del oxígeno para vivir. El proceso de la respiración al proporcionar la oxigenación, crea los llamados radicales libres en todas las células. El organismo dispone de mecanismos de defensa enzimáticos que garantizan una concentración tolerable de dichos radicales libres, ya que, en exceso, los radicales libres pueden causar daño a las células, en especial a las del sistema nervioso. Cuando la defensa antioxidante no resulta totalmente eficiente, al fallar algunos de los mecanismos naturales de protección, se incrementa la formación y acumulación de radicales libres en el organismo, se está entonces bajo los efectos del estrés oxidativo. Por tanto, el estrés oxidativo ocurre cuando hay un desequilibrio en las células debido a un aumento en los radicales libres y/o una disminución en los antioxidantes. Este desequilibrio está en la base de la mayoría de las enfermedades humanas, pero aparece especialmente presente en los trastornos que afectan la salud del sistema nervioso. Como lo señalan: Hassan W, Noreen H, Rehman S, Kamal MA, da Rocha JBT en su investigación: Association of Oxidative Stress with Neurological Disorders.
Los sistemas de defensa antioxidante, tanto enzimáticos como no enzimáticos, actúan de manera cooperativa y protegen al organismo de los riesgos que conlleva el estrés oxidativo, sin embargo, el nivel de actividad diferencial que hace que se sea más o menos capaces de mantener su funcionamiento, está determinado por polimorfismos genéticos que afectan la cinética enzimática de los enzimas claves que lo conforman. Las enzimas de Fase I transforman los productos tóxicos en formas intermedias más accesibles para la Fase II. En esta fase de activación se oxidan los tóxicos liposolubles a través de una serie de enzimas llamadas citocromos P450. El riesgo está en que estas formas intermedias son mucho más activas químicamente (por ejemplo, radicales libres) y, por lo tanto, más tóxicas, por lo que dependerán de la eficiencia de la Fase II para su neutralización y eliminación. Como enzimas de Fase I se destacan las del complejo del citocromo P450: CYP1A1, SOD.
En la Fase II participan las enzimas encargadas de neutralizar estos productos intermedios tan reactivos y tóxicos mediante diferentes vías, con el objetivo de transformarlos para favorecer su eliminación por la orina, las heces o el sudor. En esta fase, también identificada como la fase de conjugación, se reducen los productos oxidados en la fase anterior, a través de antioxidantes como los elaborados por las glutatión-transferasas (GSTs) encargadas de la conjugación con glutatión. Precisamente, se ha demostrado la asociación de una de las combinaciones genéticas que más se vincula con depleción del glutatión: el genotipo GSTM1 nulo con el TEA. Como en los resultados de Radwan K, Wu G, Banks-Word K, Rosenberger R, en su estudio: An Open-Label Case Series of Glutathione Use for Symptomatic Management in Children with Autism Spectrum Disorder.
La síntesis de glutatión puede ser promovida por una molécula conocida como N-acetilcisteína (NAC), que es un residuo de cisteína acetilada que puede servir como modulador glutamatérgico, capaz de eliminar radicales libres y es donante de cisteína para mantener el estado de GSH. Curiosamente, un metaanálisis realizado por: Lee TM, Lee KM, Lee CY, Lee HC, Tam KW, Loh EW, titulado: Effectiveness of N-acetylcysteine in autism spectrum disorders: A meta-analysis of randomized controlled trials en el 2021 mostró que la suplementación con NAC permitió aliviar la hiperactividad y la irritabilidad en casos con TEA y, además, el estudio demostró la seguridad y tolerabilidad de dicha suplementación con altas dosis de NAC en niños con TEA.
Como se ha explicado existen diversos polimorfismos genéticos que afectan a genes modificadores que determinarán que haya casos con un sistema inmune alterado donde además coexisten con otras variaciones o polimorfismos en otros genes modificadores que determinen una baja tolerancia a tóxicos. En estos casos, las interacciones entre los polimorfismos genéticos, epistasis, ejerce una fuerte influencia en el fenotipo clínico, lo que conlleva a la aparición de los diferentes subtipos que se pueden encontrar dentro de cada diagnóstico global. (Fig. 2).
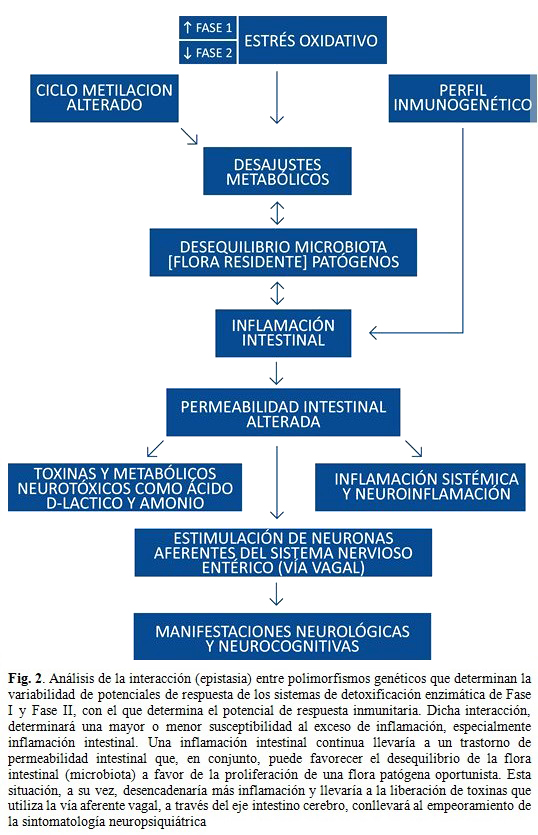
CONCLUSIONES
El análisis de la epistasia entre los polimorfismos genéticos que afectan el funcionamiento de los genes modificadores proporciona una herramienta adicional que, dentro del proceso de análisis de cada caso, con independencia del diagnóstico clínico de base, permitirá definir el subtipo clínico que mostrará dentro de la expresión del fenotipo clínico final.
Este análisis también revelará dianas moleculares sobre las que se pueden actuar para el diseño de estrategias de soporte encaminadas a mejorar el pronóstico de cada caso, al actuar de forma individual. Se trata de garantizar la mayor precisión en cada medida aplicada y en este sentido, aumentar su eficacia, pero también minimizar el impacto de efectos adversos ya que se tendrán en cuenta claves metabólicas específicas inherentes a cada caso.
En todo trastorno tan complejo por su heterogeneidad, como ocurre con los trastornos del neurodesarrollo, el análisis de la epistasia entre los genes no alélicos modificadores y el impacto o influencia que tenga su interacción con los factores no genéticos, marcará la diferencia entre aplicar una medicina estándar o una verdadera medicina de precisión.
Conflicto de intereses:
El autor declara la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: José Ignacio Lao Villadóniga.
2. Curación de datos: José Ignacio Lao Villadóniga.
3. Análisis formal: José Ignacio Lao Villadóniga.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: José Ignacio Lao Villadóniga.
6. Metodología: José Ignacio Lao Villadóniga.
7. Administración del proyecto: José Ignacio Lao Villadóniga.
8. Recursos: José Ignacio Lao Villadóniga.
9. Software: José Ignacio Lao Villadóniga.
10. Supervisión: José Ignacio Lao Villadóniga.
11. Validación: José Ignacio Lao Villadóniga.
12. Visualización: José Ignacio Lao Villadóniga.
13. Redacción del borrador original: José Ignacio Lao Villadóniga.
14. Redacción – revisión y edición: José Ignacio Lao Villadóniga.
REFERENCIAS BIBLIOGRÁFICAS
- Carter MT, Srour M, Au PB, Buhas D, Dyack S, Eaton A, et al. Genetic and metabolic investigations for neurodevelopmental disorders: position statement of the Canadian College of Medical Geneticists (CCMG). J Med Genet. 2023;60(6):523-32 [Buscar en Google Scholar]
- Lao JI. Autism Spectrum Disorders: An Intervention Approach Based on Genomic Analysis. Biol Med J. 2014;S1(S1):2 [Buscar en Google Scholar]
- Rogers JT, Weeber EJ. Reelin and apoE actions on signal transduction, synaptic function and memory formation. Neuron Glia Biol. 2009;4(3):259-70 [Buscar en Google Scholar]
- Belloy ME, Andrews SJ, Le Y, Cuccaro M, Farrer LA, Napolioni V, et al. APOE Genotype and Alzheimer Disease Risk Across Age, Sex, and Population Ancestry. JAMA Neurol. 2023;80(12):1284-94 [Buscar en Google Scholar]
- Harker SA, Al-Hassan L, Huentelman MJ, Braden BB, Lewis CR. APOE ε4-Allele in Middle-Aged and Older Autistic Adults: Associations with Verbal Learning and Memory. Int J Mol Sci. 2023;24(21):15988 [Buscar en Google Scholar]
- Stewart WF, Schwartz BS, Simon D, Kelsey K, Todd AC. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ Health Perspect. 2002;110(5):501-5 [Buscar en Google Scholar]
- Kim H, Devanand DP, Carlson S, Goldberg TE. Apolipoprotein E Genotype e2: Neuroprotection and Its Limits. Front Aging Neurosci. 2022;14(10):919712 [Buscar en Google Scholar]
- Kuroda MMM, Weck ME, Sarwark JF, Hamidullah A, Wainwright MS. Association of apolipoprotein E genotype and cerebral palsy in children. Pediatrics. 2007;119(2):306-13 [Buscar en Google Scholar]
- Shiota Y, Hirosawa T, Yoshimura Y, Tanaka S, Hasegawa C, Iwasaki S, et al. Effect of CNTNAP2 polymorphism on receptive language in children with autism spectrum disorder without language developmental delay. Neuropsychopharmacol Rep. 2022;42(3):352-5 [Buscar en Google Scholar]
- Li D, Zhang L, Bai T, Huang W, Ji GJ, Yang T, et al. Common variants of the autism-associated CNTNAP2 gene contribute to the modulatory effect of social function mediated by temporal cortex. Behav Brain Res. 2021;409(10):113319 [Buscar en Google Scholar]
- Koeda M, Watanabe A, Tsuda K, Matsumoto M, Ikeda Y, Kim W, et al. Interaction effect between handedness and CNTNAP2 polymorphism (rs7794745 genotype) on voice-specific frontotemporal activity in healthy individuals: an fMRI study. Front Behav Neurosci. 2015;9(9):87 [Buscar en Google Scholar]
- Stein MB, Yang BZ, Chavira DA, Hitchcock CA, Sung SC, Shipon E, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 is associated with increased risk for selective mutism and social anxiety-related traits. Biol Psychiatry. 2011;69(9):825-31 [Buscar en Google Scholar]
- Maina F, Panté G, Helmbacher F, Andres R, Porthin A, Davies AM, et al. Coupling Met to specific pathways results in distinct developmental outcomes. Mol Cell. 2001;7(6):1293-306 [Buscar en Google Scholar]
- Campbell DB, Sutcliffe JS, Ebert PJ, Militerni R, Bravaccio C, Trillo S, et al. A genetic variant that disrupts MET transcription is associated with autism. Proc Natl Acad Sci USA. 2006;103(45):16834-9 [Buscar en Google Scholar]
- Campbell DB, Buie TM, Winter H, Bauman M, Sutcliffe JS, Perrin JM, et al. Distinct genetic risk based on association of MET in families with co-occurring autism and gastrointestinal conditions. Pediatrics. 2009;123(3):1018-24 [Buscar en Google Scholar]
- Speranza L, di Porzio U, Viggiano D, de Donato A, Volpicelli F. Dopamine: The Neuromodulator of Long-Term Synaptic Plasticity, Reward and Movement Control. Cells. 2021;10(4):735 [Buscar en Google Scholar]
- Hain DT, Al Habbab TA, Cogan ES, Johnson HL, Law RA, Lewis DJ. Review and Meta-analysis on the Impact of the ADRA2A Variant rs1800544 on Methylphenidate Outcomes in Attention-Deficit/Hyperactivity Disorder. Biol Psychiatry Glob Open Sci. 2021;2(2):106-14 [Buscar en Google Scholar]
- Esmaiel NN, Ashaat EA, Mosaad R, Fayez A, Ibrahim M, Abdallah ZY, et al. The potential impact of COMT gene variants on dopamine regulation and phenotypic traits of ASD patients. Behav Brain Res. 2020;378(27):112272 [Buscar en Google Scholar]
- Ike KGO, Lamers SJC, Kaim S, de Boer SF, Buwalda B, Billeter JC, et al. The human neuropsychiatric risk gene Drd2 is necessary for social functioning across evolutionary distant species. Mol Psychiatry. 2024;29(2):518-28 [Buscar en Google Scholar]
- Kuc K, Bielecki M, Racicka E, Czerwinski MB, Cybulska A. The SLC6A3 gene polymorphism is related to the development of attentional functions but not to ADHD. Sci Rep. 2020;10(1):6176 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129