
Artículos originales
Pacientes jóvenes cubanos con diagnóstico clínico y bioquímico de xeroderma pigmentoso
Young Cuban Patients with a Clinical and biochemical Diagnosis of Xeroderma Pigmentosum
 ,1
Mónica Marín García,2
Anamarys Pandolfi Blanco,3
Lucía Fariñas Rodríguez,4
Paulina Araceli Lantigua Cruz,5
,1
Mónica Marín García,2
Anamarys Pandolfi Blanco,3
Lucía Fariñas Rodríguez,4
Paulina Araceli Lantigua Cruz,5
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2024-02-23 13:58:53
Aprobado: 2024-02-26 11:33:04
Correspondencia: Judith Beatriz Pupo Balboa. Centro Nacional de Genética Médica. La Habana. judith.pupo@infomed.sld.cu
RESUMEN
Objetivo: evaluar la capacidad de la reparación del ADN frente a radiación ultravioleta C y determinar la expresión de la ADN polimerasa eta en pacientes cubanos con diagnóstico presuntivo de la enfermedad.
Métodos: se realizó un estudio de serie de casos, en el que se incluyeron cuatro pacientes cubanos con diagnóstico presuntivo de xeroderma pigmentoso, establecido por una comisión médica nacional multidisciplinaria. Se evaluó la capacidad de reparación del ADN frente a radiación ultravioleta C mediante el ensayo cometa alcalino y se determinó la expresión de la polimerasa eta mediante el ensayo western blot. Se utilizaron linfocitos de sangre periférica.
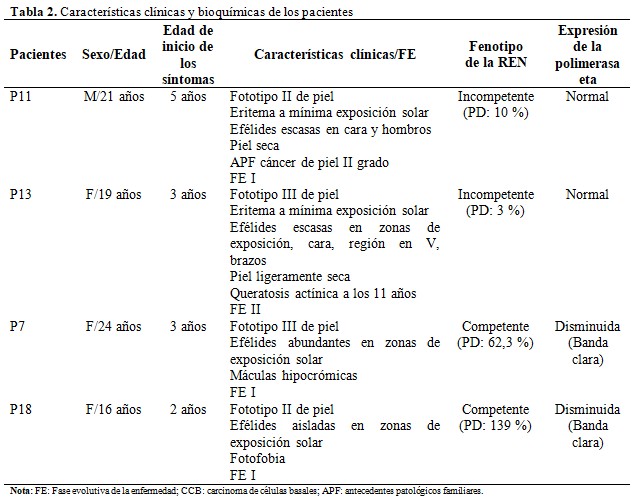
Resultados: en dos pacientes el ensayo cometa alcalino evidenció un fenotipo reparador competente (62,3 % y 139 %). Los otros dos pacientes tuvieron una reparación deficiente (10 % y 3 %), lo que sugirió que pudieran presentar alguna de las formas clásicas de la enfermedad. En los cuatro pacientes, con el western blot, se les detectó expresión de la polimerasa eta; en los dos que tuvieron la reparación normal, la banda de la proteína se observó débil, lo que indicó que pudieran presentar la forma variante de la enfermedad.
Conclusiones: la evaluación del fenotipo reparador del ADN con radiación ultravioleta C y de la expresión de la polimerasa eta en linfocitos de sangre periférica contribuye a precisar el diagnóstico clínico del XP en pacientes cubanos.
Palabras clave: xeroderma pigmentoso; reparación de escisión nucleótida; ADN polimerasa; ensayo cometa alcalino; western blot
ABSTRACT
Objective: to evaluate the capacity of deoxyribonucleic acid repair against ultraviolet C radiation and to determine the expression of eta polymerase in Cuban patients.
Methods: a case series study, including four patients with a presumptive diagnosis of xeroderma pigmentosum, established by a multidisciplinary national medical commission. The capacity of deoxyribonucleic acid repair against ultraviolet C radiation was evaluated by the alkaline comet assay and the expression of eta polymerase was determined by the western blot assay. Peripheral blood lymphocytes were used.
Results: in two patients, the alkaline comet assay showed a competent repair phenotype (62.3% and 139% respectively). The other two patients had deficient repair, suggesting the presence of one of the classic forms of the disease. In all four patients, expression of eta polymerase was detected by western blot; in the two who had normal repair, the protein band was weak, indicating that they might have the variant of the disease.
Conclusions: the evaluation of the deoxyribonucleic acid repair phenotype with ultraviolet C radiation and of the expression of eta polymerase in peripheral blood lymphocytes contributes to a precise clinical diagnosis of xeroderma pigmentosum in Cuban patients.
Key words: xeroderma pigmentosum; nucleotide excision repair; dna polymerase; alkaline comet test; western blot
INTRODUCCIóN
Las radiaciones ultravioletas (rUV) son la causa fundamental del cáncer de piel.(1) En el ADN inducen de forma directa dímeros de pirimidina y fotoproductos en regiones de pirimidinas adyacentes, que distorsionan la estructura de la doble hebra. Estas fotolesiones son eliminadas por el mecanismo de reparación del ADN por escisión de nucleótidos (REN).(1,2) En las regiones donde la REN no ha reparado el daño actúa la polimerasa eta (gen POLH) mediante la síntesis translesión, al evitar que el daño se fije como mutación durante la replicación.(2,3)
La pérdida de la función de cualquiera de estos mecanismos produce la enfermedad xeroderma pigmentoso (XP). Fue a partir de esta disfunción que se identificó la relación entre la REN y el cáncer de piel. El XP es una enfermedad genética autosómica recesiva, que ocurre en 1:1000000 de individuos en Estados Unidos y Europa y es más frecuente en Japón, el norte de África o el Oriente Medio, debido a variantes fundadoras o alta tasa de consanguinidad. Puede afectar a cualquier etnia sin distinguir el color de la piel. Los pacientes aquejados por uno u otro mecanismo presentan sensibilidad incrementada a la rUV y riesgo elevado de cáncer de piel, principalmente, lo que puede causar la muerte a edades muy tempranas. Variantes patogénicas bialélicas en ocho genes de la REN: DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, XPA y XPC, originan las formas clásicas de la enfermedad: subtipos genéticos XPE, ERCC1, XPD, XPB, XPF, XPG, XPA, XPC, respectivamente y funcionan en el reconocimiento y escisión del daño. Cuando ocurren mutaciones en el gen de la ADN polimerasa eta (POLH) se produce el XP variante (subtipo XPV). La mayoría de los casos pertenecen a los subtipos XPA, XPC, XPD o XPV. En el 25 % ocurre neurodegeneración progresiva (subtipos ERCC1, XPA, XPD, XPB, XPF, XPG). La enfermedad no tiene cura. El diagnóstico temprano, la estricta protección de las rUV y del humo de tabaco, así como el manejo óptimo, pueden mejorar la calidad y esperanza de vida de estos pacientes.(2,3,4)
El diagnóstico se basa en las manifestaciones clínicas de comienzo temprano, pero representa un reto dada la gran variabilidad clínica.(5) Existe solapamiento entre los diferentes subtipos genéticos y puede semejar características de la piel actínica y de otros desórdenes con REN afectada que cursan con fotosensibilidad. Se requieren pruebas funcionales y moleculares para confirmar el diagnóstico clínico, lo que complejiza el proceso al encontrarse habitualmente disponibles en laboratorios de investigación. Entre las primeras, la reactivación celular del huésped y la síntesis de ADN no programada, permiten analizar la capacidad de la REN frente a rUV y determinar el grupo de complementación afectado,(2,3,5) también el ensayo cometa alcalino permite evaluar el funcionamiento de este mecanismo frente a rUV-C con resultados más rápidos que las técnicas anteriores, al no requerir cultivo de células.(6,7) Las otras pruebas incluyen: el análisis de secuencias del gen afectado para identificar la mutación presente; estudios de expresión del ARN mensajero y a nivel de proteínas, ya que la mayoría de las mutaciones que causan el XP resultan en niveles reducidos o ausentes de la proteína mutada.(2,8) Es posible detectar pacientes XPA, XPC y XPV por inmunohistoquímica o western blot con anticuerpos específicos, mientras que en pacientes XPB y XPD son menos apropiados, al no producirse una reducción significativa de las proteínas.(2)
En Cuba, desde el 2003, una comisión médica nacional multidisciplinaria designada por el Ministerio de Salud Pública evalúa a los pacientes con manifestaciones clínicas sugestivas de XP. En el país no están disponibles las técnicas moleculares para la identificación de la mutación presente y confirmación diagnóstica, pero se ha aplicado el ensayo cometa alcalino para evaluar el funcionamiento de la REN frente a rUV-C en pacientes evaluados por la comisión. Los resultados demostraron la utilidad de esta técnica para delinear el fenotipo reparador de estos pacientes, y determinar que la mayoría de ellos no clasificaban como XP clásico, sino que podrían presentar la forma variante de la enfermedad o tratarse de una marcada sensibilidad a las radiaciones solares debido a fototipos bajos de piel.(9,10) Como parte de la estrategia diagnóstica y para continuar el estudio de pacientes identificados por la comisión fue posible introducir el ensayo western blot para la detección de la ADN polimerasa eta (POLH/XPV). Por lo que en la investigación se evaluó la capacidad de la reparación del ADN frente a la radiación ultravioleta C y se determinó la expresión de la polimerasa eta en pacientes cubanos con diagnóstico presuntivo de la enfermedad.
MéTODOS
Se realizó un estudio de serie de casos de enero a junio del año 2018 en el Centro Nacional de Genética Médica (CNGM). Incluyó a cuatro pacientes con diagnóstico presuntivo de xeroderma pigmentoso, previamente evaluados por la comisión nacional multidisciplinaria de la enfermedad y con seguimiento en la consulta de Genética Clínica de Referencia Nacional del Hospital Pediátrico Universitario Juan Manuel Márquez. Se aplicó un algoritmo de trabajo para contribuir al diagnóstico de la enfermedad, fundamentado en los hallazgos clínicos, la historia natural de la enfermedad, la evaluación de la capacidad de reparación por escisión de nucleótidos frente a rUV-C y la determinación de la expresión de la polimerasa eta. Se realizó una comparación de pacientes XPV de la literatura con los pacientes del estudio que presentaron características clínico-bioquímicas de XP variante. Se tuvo en cuenta que los parámetros hematológicos y de bioquímica sanguínea (hemoglobina, leucograma, glicemia, creatinina, ácido úrico y actividades enzimáticas hepáticas) estuvieran normales. Se excluyeron, el embarazo, la lactancia, las enfermedades infecciosas y crónicas descompensadas, la exposición a rayos X y rayos gamma y la ingesta de medicamentos 30 días previos a la toma de la muestra. Para la determinación de los marcadores de interés se utilizó sangre periférica, 3 mL colectados en tubo con EDTA y 2 mL en tubo seco, que fueron procesados antes de las 4 horas de extraídas. (Fig.1).
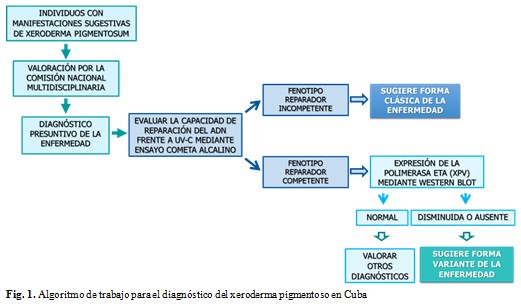
Aislamiento de los linfocitos de la sangre
Los linfocitos aislados de un mL de sangre, según procedimiento habitual(11) se re-suspendieron en un medio de cultivo Roswell Park Memorial Institute (RPMI) (por sus siglas en inglés) 1640 frío a 1x105 células/mL, para realizar el ensayo cometa alcalino. Las células aisladas de dos mL de sangre se utilizaron en la obtención de los lisados celulares.
Capacidad de reparación frente a UV-C mediante ensayo cometa alcalino
Se indujo daño en el ADN con rUV-C (254 nm) para evaluar la eficiencia de la REN.(7,12) El tratamiento de las células y los tiempos de incubación de la cinética de reparación tuvieron modificaciones respecto a lo descrito previamente por el grupo de trabajo.(11) En el procedimiento, la suspensión celular de linfocitos en RPMI 1640 frío se irradió seis segundos en placas de cultivo de 35 mm sobre hielo con una lámpara UV Chromato-Vue C-70G, a dosis de 20 J/m2. La intensidad de la irradiación se midió con un foto-radiómetro HD2 102.2 con sonda UV-C. Los linfocitos irradiados se mezclaron con agarosa de bajo punto de fusión al 0,5 % y se aplicaron sobre duplicados de láminas portaobjetos.(11) A continuación, las láminas se incubaron en un medio de cultivo Quantum PBL a 37ºC y 5 % CO2 durante 0, 15, 45, 60 y 240 minutos. Continuó la lisis, el desenrollamiento, la desnaturalización del ADN, la electroforesis alcalina y la cuantificación del daño del ADN en unidades arbitrarias (UA) de los núcleos teñidos con nitrato de plata, según el procedimiento.(11)
La capacidad de reparación del ADN frente a la rUV-C (fenotipo de la REN) se determinó como el porciento de la diferencia (PD) entre los 0 y 45 minutos posteriores a la radiación. La REN se consideró competente cuando la reparación mostró una eficiencia mayor del 20 %. (9,10) Se utilizó como referencia la cinética de sujetos aparentemente sanos, controles del laboratorio, con igual diseño experimental.
Detección de la polimerasa eta (POLH/XPV) mediante western blot
Obtención de lisados celulares o extractos proteicos totales: las células aisladas de dos mL de sangre se re-suspendieron en 75 µL de tampón fosfato salino frío, (PBS) (por sus siglas en inglés), suplementado con una mezcla de inhibidores de proteasas, (número de catálogo: P8340, Sigma-Alcrich), orto vanadato de sodio (número de catálogo: S6508, Sigma-Alcrich) y fenilmetano sulfonil fluoruro (número de catálogo: P7626, Sigma-Alcrich). Se añadieron 75 µL de tampón Laemmli 2x (número de catálogo: sc-286962, Santa Cruz Biotechnology, USA), se agitó en vórtex durante 30 segundos y los extractos se hirvieron durante 5 minutos de 95 a 100ºC.
La electroforesis de proteínas se realizó en geles de poliacrilamida al 10 %, de 1 mm de grosor (peso molecular de XPV 78.4KDa; OMIM 603968) a 100 V durante 1 h y 30 minutos, con tampón Tris-Borato-SDS pH 8.64 en la cámara superior y tampón Tris-HCl pH 9.18 en la cámara inferior. Se aplicaron 25 µL de los extractos proteicos, equivalentes a 35 µg de proteína total y 10 µL de marcadores de peso molecular de amplio rango (número de catálogo: sc-2361, Santa Cruz Biotechnology, USA)
Se realizó la transferencia proteica por difusión pasiva a una membrana de nitrocelulosa de 0,45 µm (número de catálogo: sc-3724, Santa Cruz Biotechnology, Inc.) con tampón Tris-HCL, metanol al 20 % y SDS al 0,02 %, durante toda la noche a temperatura ambiente. El gel y la membrana se lavaron 20 minutos con este tampón. El gel, la membrana y una torre de 5 cm de papeles de filtro de dimensiones iguales se colocaron sobre un soporte dentro de una bandeja que contenía tampón de transferencia. Se utilizó un puente de papel de filtro, cuyos extremos quedaron embebidos en el tampón y se colocó un peso de 1 kg sobre el sistema. La transferencia de las proteínas se comprobó tiñendo las membranas de nitrocelulosa con solución de rojo Ponceau y ácido acético al 5 %.
Fase inmunológica para la detección de la proteína XPV: el bloqueo de la membrana se realizó en tampón Tris-salino (TBS), Tween 20 al 0,05 % y albúmina de suero bovino (BSA) al 1 % durante 1 h. Se incubó con el anticuerpo primario monoclonal de ratón POL H (B-7), número de catálogo sc-17770 (dilución 1:50), en tampón de bloqueo toda la noche a 17ºC y agitación lenta. La membrana se lavó 5 minutos por cuatro veces con tampón TBS-Tween 20 al 0,05 %. Se incubó con el anticuerpo secundario anti IgG Fab específico de ratón conjugado con fosfatasa alcalina, número de catálogo A2179, Sigma-Alcrich (dilución 1:5000), en tampón de bloqueo y agitación lenta, 1h a temperatura ambiente. La membrana se lavó de forma similar y se realizó el revelado con BCIP/NBT (número de catálogo B3679, Sigma-Alcrich). Como control de carga de las muestras se utilizó la detección de la beta actina con el anticuerpo monoclonal de ratón, número de catálogo A2228, Clone AC-74, Sigma-Alcrich, a 0,5 µg/m y el referido anticuerpo secundario en iguales condiciones de trabajo.
Al daño cuantificado en UA en cada tiempo de incubación le fue restado el daño basal medido a los 0 minutos. La eficiencia de reparación se evaluó a los 45 minutos de incubación. Como valores de referencia del laboratorio se utilizó una cinética de reparación en sujetos aparentemente sanos en el rango de edades de los pacientes y con igual diseño experimental. De acuerdo a esta cinética, el daño promedio de ADN fue mayor a los 45 minutos. El tercer cuartil del daño del ADN en este punto (158,5 UA) se empleó como punto de corte; y el valor promedio de la actividad de reconocimiento y escisión de la REN (100,4 % ± 26), se consideró como el 100 %. Se realizó la evaluación cualitativa de la expresión de la proteína XPV: normal, cuando la banda de la proteína fue del tamaño molecular esperado y de intensidad similar a la del control positivo (C+); disminuida: cuando la banda de la proteína fue del tamaño molecular esperado, pero de intensidad menor a la del C+ (banda clara); ausente: cuando no se detectó la banda de la proteína del tamaño molecular esperado.
Esta investigación siguió los principios éticos para las investigaciones médicas en seres humanos establecidos en la Declaración de Helsinki y fue aprobada por el Comité de Ética de la Investigación y el Consejo Científico del Centro Nacional de Genética Médica. En el proceso de consentimiento informado se comunicó a los participantes los potenciales riesgos, beneficios y todos sus derechos. Los pacientes expresaron la voluntariedad de participar mediante la firma del consentimiento informado (los tutores, en el caso de los menores de edad). Se garantizó la confidencialidad de la información.
RESULTADOS
Cinética de reparación frente a rUV-C y fenotipo reparador del ADN
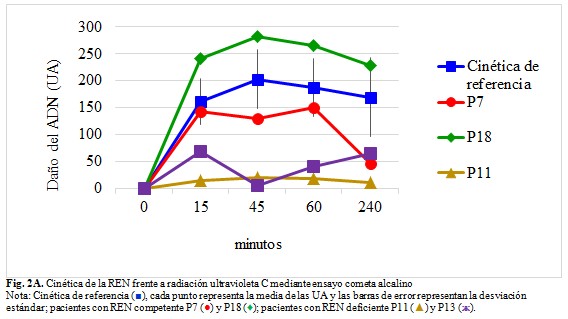
Tres pacientes presentaron el daño del ADN por debajo del punto de corte (158,5 UA, punto de corte de los valores de referencia del laboratorio, tercer cuartil del daño del ADN a los 45 minutos de incubación), sin embargo, en dos de ellos, la capacidad de reparación (PD a los 45 minutos) fue mayor del 20 % del valor promedio de referencia del laboratorio, considerado como el 100 % de la actividad de reconocimiento y escisión de la REN (100,4 % ± 26), por lo que el fenotipo de la REN se clasifica como competente. Los otros dos pacientes mostraron una cinética de reparación con una curva plana y una reparación incompetente, al tener un PD menor del 20 % del valor promedio de referencia del laboratorio. (Tabla 1).
Expresión de la polimerasa eta (POLH/XPV)

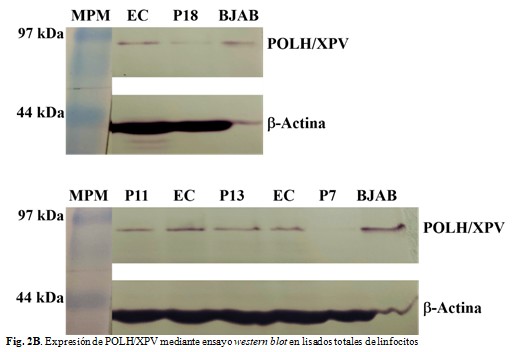
En todos los pacientes se detectó la proteína XPV del tamaño esperado, aunque en los dos que mostraron una REN competente por ensayo cometa, la banda de la proteína se observó de intensidad débil en P7 y P18. En los dos individuos con REN deficiente (P11 y P13) la evaluación cualitativa de la señal de la proteína se consideró normal.
Para el análisis bioquímico de los pacientes se realizaron:
A: Cinética de la REN frente a radiación ultravioleta C, mediante ensayo cometa alcalino. Al daño del ADN cuantificado en unidades arbitrarias (UA) en cada tiempo de incubación le fue restado el daño basal medido a los 0 minutos de incubación. (Fig. 2A).
B: Expresión de POLH/XPV mediante ensayo western blot en lisados totales de linfocitos. Como control positivo se utilizaron extractos proteicos de la línea celular BJAB (número de catálogo sc-2145) y extractos controles (EC). Expresión normal: P11 y P13. Expresión disminuida: P18 y P7. (Fig. 2B).
De la revisión de la historia clínica genética, el cuestionario y los ensayos de laboratorio se obtuvieron datos para caracterizar los pacientes según el sexo, la edad, el momento de inicio de los síntomas, características clínicas y otros que mostraron un predominio del sexo femenino y edades comprendidas entre los 16 y 21 años, con una edad mínima de inicio de los síntomas a los 2 años de edad. En el caso de dos pacientes la capacidad de reparación del ADN frente a rUV-C determinada mediante ensayo cometa alcalino mostró un fenotipo de la REN incompetente, con valores normales de la expresión de la polimerasa eta determinado por el ensayo western blot. De igual forma los otros dos pacientes presentaron valores consecuentes de fenotipo de la REN competente y expresión disminuida de la polimerasa eta para los dos ensayos realizados. (Tabla 2).
Comparación de los pacientes del estudio con pacientes XPV de la literatura
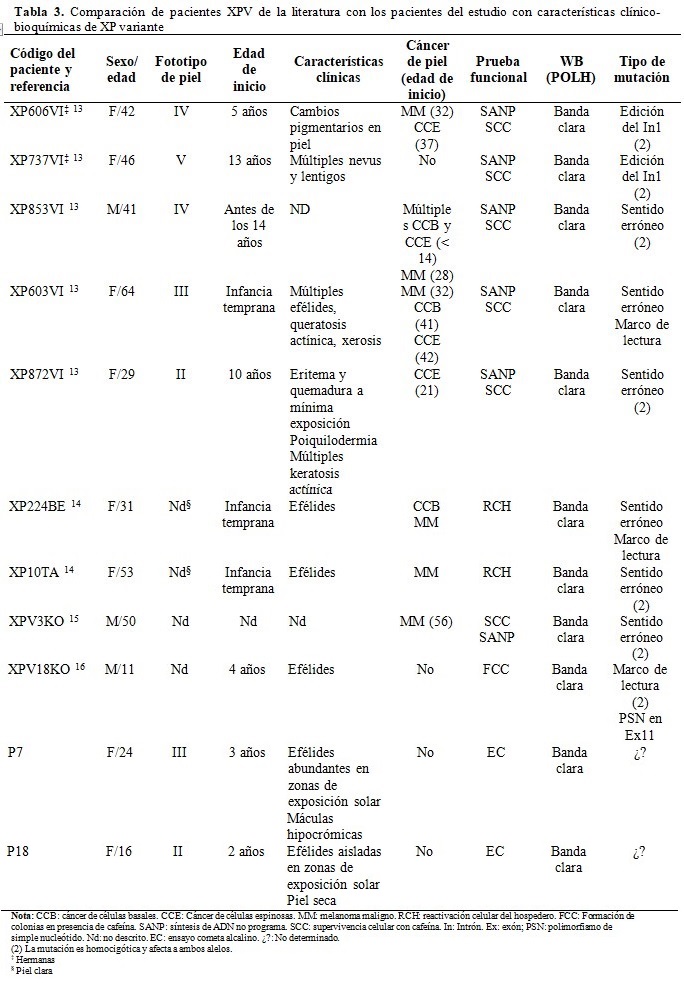
Se realizó una comparación de los pacientes que tuvieron la REN normal y la banda de la POLH disminuida con pacientes XPV reportados en la literatura que, por inmunodetección, mostraron bandas claras de la proteína del tamaño esperado. Los pacientes descritos tuvieron una clínica variable, desde fenotipos leves, sin presencia de cáncer de piel (XP737VI, XPV18KO), hasta aquellos más graves con múltiples lesiones tumorales en la piel a edades tempranas (XP853VI, XP872VI). La generalidad tuvo un inicio de los síntomas en los primeros años de la infancia, excepto el paciente XPV3KO de la referencia 15, del que no se describió la edad de inicio, el fototipo de piel ni las características clínicas; del mismo modo, la edad de inicio del cáncer de piel en los pacientes de la referencia 14 no se informó.
En comparación, los pacientes P7 y P18, también, tuvieron los primeros síntomas en la infancia temprana y una presentación leve de los rasgos clínicos, sin la presentación de cáncer de piel. El análisis de las mutaciones realizado mediante secuenciación del ADN genómico, comprobado por secuenciación de Sanger, identificó en la mayoría de los pacientes referidos en la literatura, mutaciones con sentido erróneo en homocigosis o heterocigosis. En el caso de los pacientes de esta investigación no fue posible realizar estos estudios para identificar la mutación presente. (Tabla 3).
DISCUSIóN
Fenotipo reparador del ADN frente a rUV-C y expresión de la ADN polimerasa eta (POLH XPV) en los pacientes
De acuerdo con el algoritmo de trabajo para contribuir al diagnóstico de la enfermedad, se determinó la capacidad de reparación por escisión de nucleótidos frente a la rUV-C por ensayo cometa alcalino. El ensayo cometa alcalino con rUV-C se ha utilizado como prueba funcional para determinar el fenotipo reparador del ADN en pacientes con mutaciones en genes de la REN.(6,7) La fase inicial de este mecanismo, de reconocimiento y escisión del daño, es donde participan las proteínas XPA a XPG y ERCC1, que, de estar mutadas, causan las formas clásicas de la enfermedad. Como consecuencia de la actividad de estas enzimas, el ensayo cometa alcalino detecta un mayor número de roturas del ADN, lo que evidencia el funcionamiento de la REN.(7) En la investigación realizada, con similitud a resultados anteriores del grupo de trabajo,(10) esta fase se correspondió con el intervalo entre 0 y 45 minutos de incubación posterior a la irradiación; tiempo donde se cuantificó el máximo daño.
El fenotipo reparador deficiente que exhibieron los pacientes P11 y P13 señaló que podrían presentar algunas de las formas clásicas de la enfermedad. Resultados similares por ensayo cometa alcalino con rUV-C fueron descritos anteriormente en pacientes cubanos, aunque contrastó que el 70,6 % tuvo la REN afectada.(9) En contraste, el fenotipo reparador eficiente observado en los pacientes P7 y P18, indica que no son pacientes XP clásicos, sino que pudieran presentar XP variante u otro diagnóstico en el que la REN no esté afectada. Estudios previos en individuos con manifestaciones clínicas de hipersensibilidad a la radiación solar, realizados por el grupo de trabajo, mostraron resultados similares.(10,17)
En virtud de la utilidad de determinar la expresión de la polimerasa eta mediante WB para el diagnóstico de la forma XP variante,(2,8) se determinó la expresión de XPV como parte del algoritmo de trabajo para contribuir al diagnóstico de la enfermedad. La presencia de una banda de intensidad débil en los pacientes con REN competente, que indicó una expresión disminuida de la proteína, sugirió que podrían ser XP variante. Aunque se requiere identificar el defecto molecular, estos resultados son congruentes con varios estudios de pacientes XP variante por medio de inmunoprecipitación y WB en fibroblastos y células linfoblastoides, en los que se observaron bandas claras de POLH,(13,14,15,16) además, de la ausencia de la proteína.(18) Los dos pacientes con REN incompetente mostraron una expresión normal de la proteína, lo que permitió verificar los resultados obtenidos en el ensayo cometa alcalino y utilizarlos, además, como controles positivos del WB.(13)
Pacientes con fenotipo reparador incompetente
Si se tiene en cuenta la historia natural de la enfermedad, la evolución clínica, la prevalencia de cada grupo de XP a nivel mundial cabría plantear que estos pacientes fueran XPC. Este subtipo genético es el segundo en frecuencia.(1,4) Los pacientes XPC tienen conservada la vía de la REN acoplada a la transcripción, con actividad residual de reparación (determinada por síntesis no programada de ADN) del 10 al 20 % y hasta del 40 % en individuos con diagnóstico tardío.(4,5) En el presente estudio los pacientes con la REN deficiente tuvieron reparaciones por debajo de estos valores, aunque por ensayo cometa alcalino.
El fenotipo clínico de individuos XPC presenta efélides graduales sin una respuesta severa de quemadura a la mínima exposición solar y el desarrollo temprano de cáncer de piel. No suele existir afectación neurológica,(5,8) y se observa una enfermedad ocular progresiva más severa con respecto a otros grupos de complementación.(5) De manera similar, los pacientes del presente trabajo con REN deficiente iniciaron los síntomas en la infancia temprana, con efélides y signos de fotosensibilidad, sin reacción exagerada a la mínima exposición y no presentaron manifestaciones neurológicas ni oftalmológicas.
En los pacientes XPC la mutación presente y el fototipo de piel en relación con la exposición a las rUV, definen la intensidad de los síntomas cutáneos. En individuos con la misma mutación y con fototipos diferentes (alto y bajo), se ha reportado una presentación más severa con cáncer de piel y cambios pigmentarios en el sujeto de piel clara y antecedentes de no adherencia a la protección de las rUV, no obstante, pacientes XPC con diagnóstico al nacimiento, que se han protegido de las rUV no han desarrollado cáncer de piel.(4) De forma comparable, en los pacientes en análisis, los síntomas cutáneos de fotodaño no han avanzado, después de haberse protegido de la rUV una vez diagnosticados clínicamente. Al momento del estudio se clasificaron en las etapas evolutivas eritemato-pigmentaria y atrófico-telangiectásica, con fototipos de piel bajo e intermedio, respectivamente. La REN deficiente en ellos pudo causar que el paciente P13 desarrollara queratosis actínica a temprana edad, aun cuando el fototipo de piel no fuera tan bajo, mientras que una susceptibilidad genética agregada fue apreciada en el paciente P11, ante la presencia de antecedentes familiares de cáncer de piel, factor que puede incrementar su riesgo.(19) Por otra parte, los pacientes XPC, también, pueden desarrollar cáncer interno. Se ha descrito la presencia de lesiones tumorales intracraneales en series de pacientes.(5) Este hallazgo parece estar relacionado con la participación de XPC en la eliminación del daño oxidativo del ADN, además de intervenir en la reparación del daño directo causado por la rUV. Su ausencia induce cambios del estado redox en la mitocondria y la acumulación de daño oxidativo del ADN en este organelo se ha asociado directamente con el desarrollo de tumores en estos pacientes.(20) Hasta el momento de esta investigación, los pacientes P11 y P13 no habían presentado este tipo de complicación.
Para confirmar el diagnóstico en estos dos individuos estudiados, en investigaciones futuras, se pudiera determinar, primeramente, la expresión de la proteína XPC mediante WB(2) y de acuerdo al resultado obtenido detectar las mutaciones presentes.
Pacientes con fenotipo reparador competente y expresión disminuida de la polimerasa eta
El análisis comparativo de los pacientes de la actual investigación con REN normal y expresión disminuida de la POLH con pacientes XPV reportados en la literatura que tuvieron un resultado análogo de la expresión de la proteína,(13,14,15,16) reveló una similitud con los fenotipos leves de los casos de la literatura. Esto podría deberse a que con el uso de medios de protección contra la rUV estos pacientes, incluidos P7 y P18, mantuvieron un cuadro clínico sin cambios significativos y evitaron la aparición de cáncer de piel,(4,16) hasta el momento que entraron a la investigación.
De acuerdo al tipo de mutación que predominó en la mayoría de los pacientes XPV referidos en la literatura, las mutaciones con sentido erróneo, aunque son menos frecuentes, a menudo presentan alguna actividad residual. Por tanto, el inicio de los síntomas de la enfermedad podría ser más tardío y la progresión depender del fototipo de piel y los niveles de exposición a la rUV.(16) Desde el punto de vista de la proteína, este tipo de mutación conservó el tamaño de la proteína, pero causó disminución de su expresión a niveles casi indetectables por inmunodetección; probablemente, debido a cambios en la conformación que desestabilizan su estructura y la hacen más inestable.(15,17)
Sobre la base de lo anterior y los resultados del WB podría considerarse la posibilidad de que los individuos P7 y P18 con características clínico-bioquímicas de XP variante, pudieran tener una mutación con sentido erróneo que origine que la POLH se observe disminuida, no obstante, el diagnóstico confirmatorio únicamente se realizaría si se detectan las mutaciones en el gen POLH. Al mismo tiempo, habría que descartar que estos pacientes portaran algún polimorfismo del gen que redujera los niveles de expresión y de actividad de la proteína. Glick, en su artículo: Mutations in DNA polymerase eta are not detected in squamous cell carcinoma of the skin, publicado en 2006 llegó a la conclusión de que: ni las mutaciones, ni los polimorfismos en las regiones codificantes de POLH son necesarios para la generación del carcinoma de células escamosas de la piel humana.
La investigación realizada tuvo como limitante que no se determinó el grupo de complementación implicado en los pacientes con REN deficiente y no se evaluó la sensibilidad post UV frente a la cafeína en los dos pacientes con expresión disminuida de POLH. Estos criterios bioquímicos, unidos a la clínica, son necesarios para definir un paciente XP.(8) Del mismo modo, no se realizaron estudios de secuencia del ADN que identificaran las posibles variantes patogénicas o polimórficas; estos estudios no estaban disponibles en el país, no obstante, esta investigación constituyó un paso de avance, aunque no definitivo, en el diagnóstico del XP en pacientes cubanos. Los marcadores bioquímicos utilizados constituyen una herramienta útil menos costosa, que orienta el diagnóstico. Finalmente se destaca que el diagnóstico clínico temprano, la fotoprotección, como mecanismo principal de tratamiento y el seguimiento clínico continuo, previenen el desarrollo de lesiones malignas en estos pacientes.
El estudio permitió la evaluación del fenotipo reparador del ADN con radiación ultravioleta C mediante el ensayo cometa alcalino y de la expresión de la polimerasa eta en linfocitos de sangre mediante el ensayo western blot como métodos que contribuyen a precisar el diagnóstico clínico del XP en pacientes cubanos.
AGRADECIMIENTOS
Los autores agradecen a todos los pacientes, sujetos sanos y tutores por acceder a participar en la investigación, así como a la Comisión Nacional Multidisciplinaria y a los dermatólogos que evaluaron a estos pacientes.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Roles de autoría:
1. Conceptualización: Judith Beatriz Pupo Balboa, Mónica Marín García, Paulina Araceli Lantigua Cruz.
2. Curación de datos: Judith Beatriz Pupo Balboa.
3. Análisis formal: Judith Beatriz Pupo Balboa, Mónica Marín García, Paulina Araceli Lantigua Cruz.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: Judith Beatriz Pupo Balboa, Mónica Marín García, Anamarys Pandolfi Blanco, Lucía Fariñas Rodríguez, Paulina Araceli Lantigua Cruz.
6. Metodología: Judith Beatriz Pupo Balboa, Mónica Marín García, Anamarys Pandolfi Blanco, Lucía Fariñas Rodríguez, Paulina Araceli Lantigua Cruz.
7. Administración del proyecto: Judith Beatriz Pupo Balboa.
8. Recursos: Judith Beatriz Pupo Balboa.
9. Software: Judith Beatriz Pupo Balboa.
10. Supervisión: Judith Beatriz Pupo Balboa.
11. Validación: Judith Beatriz Pupo Balboa, Mónica Marín García, Anamarys Pandolfi Blanco, Lucía Fariñas Rodríguez, Paulina Araceli Lantigua Cruz.
12. Visualización: Judith Beatriz Pupo Balboa.
13. Redacción del borrador original: Judith Beatriz Pupo Balboa, Mónica Marín García.
14. Redacción, revisión y edición: Judith Beatriz Pupo Balboa, Mónica Marín García, Paulina Araceli Lantigua Cruz, Anamarys Pandolfi Blanco, Lucía Fariñas Rodríguez.
REFERENCIAS BIBLIOGRÁFICAS
- Martens MC, Steffen E, Boeckmann L. Xeroderma Pigmentosum: Gene Variants and Splice Variants. Genes. 2021;12(8):1173 [Buscar en Google Scholar]
- Cleaver JE. Diagnosis of Xeroderma Pigmentosum and Related DNA Repair-Deficient Cutaneous Diseases. Curr Med Lit Dermatol. 2008;13(2):41-8 [Buscar en Google Scholar]
- Kraemer KH, DiGiovanna JJ, Tamura D. Xeroderma Pigmentosum [Internet]. Seattle: University of Washington; 2022 [citado 24 Mar 2023]. Disponible en: https://www.ncbi.nlm.nih.gov/books/NBK1397/ [Buscar en Google Scholar]
- Lehmann J, Seebode C, Martens MC, Steffen E. Xeroderma Pigmentosum. Facts and Perspectives. Anticancer Research. 2018;38(2):1159-64 [Buscar en Google Scholar]
- Fassihi H, Sethib M, Fawcett H, Wing J, Chandlerd N, Mohammed S, et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpected heterogeneity dependent on the precise molecular defect. PNAS. 2016;113(9):E1236-45 [Buscar en Google Scholar]
- Theron T, Fousteri MI, Volker M, Harries LW, Botta E, Stefanini M et al. Transcription-Associated Breaks in Xeroderma Pigmentosum Group D Cells from Patients with Combined Features of Xeroderma Pigmentosum and Cockayne Syndrome. Mol Cell Bio. 2005;25(18):8368-78 [Buscar en Google Scholar]
- Alapetite C, Benoit A, Moustacchi E, Sarasin A. The comet assay as a repair test for prenatal diagnosis of Xeroderma pigmentosum and trichothiodystrophy. J Invest Dermatol. 1997;108(2):154-9 [Buscar en Google Scholar]
- Moriwaki S, Kanda F, Hayashi M, Yamashita D, Sakai Y, Nishigori Ch. Xeroderma pigmentosum clinical practice guidelines. J Dermatol. 2017;44(10):1087-96 [Buscar en Google Scholar]
- Roblejo H, Cuétara E, Acosta T, Lantigua A, Gutiérrez R, Pupo J, et al. Descripción clínica y capacidad de reparación del ADN por escisión de nucleótidos en pacientes cubanos con Xeroderma Pigmentoso. Rev Cubana Genet Comunit [Internet]. 2009 [citado 24 Mar 2023];3(1):[aprox. 6p]. Disponible en: https://bvs.sld.cu/revistas/rcgc/v3n1/rcgc080109.htm [Buscar en Google Scholar]
- Pupo JB, Gutiérrez R, Lantigua A, Suárez NL, Acosta T, Pandolfi A, et al. A. Fenotipo de la reparación por escisión de nucleótidos en pacientes cubanos con hipersensibilidad al sol. Rev Cubana Pediatr [Internet]. 2014 [citado 24 Mar 2023];86(2):[aprox. 12p]. Disponible en: https://scielo.sld.cu/pdf/ped/v86n2/ped02214.pdf [Buscar en Google Scholar]
- Pupo JB, Gutiérrez R, Pandolfi A, Cásido M, Valdés L, de Armas A. Daño basal del ADN en un grupo de individuos cubanos sanos mediante ensayo cometa. Acta Bioquím Clín Latinoam [Internet]. 2014 [citado 24 Mar 2023];48(3):[aprox. 5p]. Disponible en: https://www.scielo.org.ar/pdf/abcl/v48n3/v48n3a10.pdf [Buscar en Google Scholar]
- Cipollini M, He J, Rossi P, Baronti F, Micheli A, Rossi A, et al. Can individual repair kinetics of UVC-induced DNA damage in human lymphocytes be assessed through the comet assay?. Mutation Research. 2006;601(1-2):150-61 [Buscar en Google Scholar]
- Opletalova K, Bourillon A, Yang W, Pouvelle C, Armier J, Despras E, et al. Correlation of phenotype/genotype in a cohort of 23 xeroderma pigmentosum-variant patients reveals 12 new disease-causing POLH mutations. Hum Mutat. 2014;35(1):117-28 [Buscar en Google Scholar]
- Inui H, Oh KS, Nadem C, Ueda T, Khan AM. Xeroderma Pigmentosum-Variant Patients from America, Europe, and Asia. J Invest Dermatol. 2008;128(8):2055-68 [Buscar en Google Scholar]
- Tanioka M, Masaki T, Ono R, Nagano T, Otoshi E, Matsumura Y, et al. Molecular Analysis of DNA Polymerase Eta Gene in Japanese Patients Diagnosed as Xeroderma Pigmentosum Variant Type. Journal of Investigative Dermatology. 2007;127(7):1745-51 [Buscar en Google Scholar]
- Ono R, Masaki T, Takeuchi T, Shimizu A, Tanioka M, Kambe N, et al. Three school‐age cases of xeroderma pigmentosum variant type. Photodermatol Photoimmunol Photomed. 2013;29(3):132-9 [Buscar en Google Scholar]
- Pupo JB, Acosta T, Gutiérrez R, Riverón G, Lantigua A, Pandolfi A, et al. Reparación por escisión de nucleótidos y estado redox en pacientes cubanos con hipersensibilidad a la radiación solar. Rev Cubana Genet Comunit [Internet]. 2016 [citado 24 Mar 2023];10(2):[aprox. 9p]. Disponible en: https://bvs.sld.cu/revistas/rcgc/v10n2/rcgc050216.html [Buscar en Google Scholar]
- De Palma A, Morren MA, Ged C, Pouvelle C, Taïeb A, Aoufouchi S, et al. Diagnosis of Xeroderma pigmentosum variant in a young patient with two novel mutations in the POLH gene. Am J Med Genet. 2017;173(9):2511-6 [Buscar en Google Scholar]
- Berlin N, Cartmel B, Leffell D, Bale A, Mayne S, Ferrucci L. Family History of Skin Cancer is Associated with Early-Onset Basal Cell Carcinoma Independent of MC1R Genotype. Cancer Epidemiology. 2015;39(6):1078-83 [Buscar en Google Scholar]
- Mori MP, Costa RA, Soltys DT, Freire TS, Rossato FA, Amigo I, et al. Lack of XPC leads to a shift between respiratory complexes I and II but sensitizes cells to mitochondrial stress. Scientific Reports. 2017;7(1):155 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129