
Artículos originales
Implementación de una metodología para la detección de marcadores bioquímicos en la tirosinemia tipo 1
Implementation of a Methodology for the Detection of Biochemical Markers in Type 1 Tyrosinemia

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2023-08-15 15:13:40
Aprobado: 2023-11-13 12:35:46
Correspondencia: Iovana Fuentes Cortés. Centro Nacional de Genética Médica. La Habana. ifuentes@cngen.sld.cu
RESUMEN
Objetivo: implementar una metodología de trabajo para la detección de metabolitos marcadores de la tirosinemia tipo 1.
Método: se realizó un estudio descriptivo y transversal en una serie de casos evaluados en el período comprendido entre enero del 2021 a febrero del 2023. Como examen inicial se realizaron las pruebas cualitativas de α nitroso beta naftol y 2,4 dinitrofenilhidracina para la identificación de la tirosina y los α cetoácidos respectivamente. Luego se realizó el método HPLC para la cuantificación de la tirosina y CG-EM para la determinación del perfil cromatográfico en orina como técnicas confirmatorias.
Resultados: las pruebas cualitativas resultaron positivas para la tirosina y sus metabolitos, así como, para los alfa cetoácidos. El perfil de ácidos orgánicos mostró excreción elevada de los metabolitos marcadores de la enfermedad en 8 casos, a los cuales se les fue realizada la cuantificación de la tirosina que resultaron positivos de padecer la enfermedad.
Conclusiones: la implementación de la metodología resultó ser una herramienta valiosa en el diagnóstico temprano de la enfermedad.
Palabras clave: marcadores bioquímicos; deficiencia de la enzima; metodología
ABSTRACT
Objective: to implement a work methodology for the detection of marker metabolites of type 1 tyrosinemia.
Method: a descriptive and cross-sectional study was carried out in a series of cases evaluated in the period from January 2021 to February 2023. As an initial examination, qualitative tests of α nitroso beta naphthol and 2,4 dinitrophenylhydrazine were carried out for the identification of tyrosine and α keto acids respectively. Then, the HPLC method was performed for the quantification of tyrosine and GC-MS for the determination of the chromatographic profile in urine as confirmatory techniques.
Results: qualitative tests were positive for tyrosine and its metabolites, as well as for alpha keto acids. The organic acid profile showed elevated excretion of the disease marker metabolites in 8 cases, in which tyrosine quantification was performed, which were positive for suffering from the disease.
Conclusions: the implementation of the methodology turned out to be a valuable tool in the early diagnosis of the disease.
Key words: biochemical markers; enzyme deficiency; methodology
INTRODUCCIÓN
La tirosinemia tipo 1 (TT1) (OMIM276700)(1) también llamada tirosinemia hepatorrenal es un error innato del metabolismo (EIM) de los aminoácidos con una incidencia de aproximadamente 1:100 000 nacimientos.(2) Se considera un padecimiento de herencia autosómica recesiva y se debe a un defecto de la enzima fumarilacetoacetato hidrolasa (FAH).(3) Bioquímicamente se caracteriza por hipertirosinemia (HT), concentraciones aumentadas en orina del aminoácido tirosina, ácidos 4-hidroxifenilderivados y niveles elevados de succinilacetona (SA) en plasma y orina, metabolito este último patognomónico de la enfermedad.(4,5)
Como primera aproximación para su diagnóstico se utilizan las pruebas químicas cualitativas (PQC), que se basan en la identificación de grupos funcionales por reacciones colorimétricas o turbidimétricas. La prueba colorimétrica α nitroso β naftol, permite la detección de los metabolitos productos de la tirosinemia: tirosina, 4-hidroxifenilacetato y el 4-hidroxifenil-lactato), dando lugar a la formación de un complejo rojo-anaranjado. Esta entidad, puede resultar positiva ante la prueba del tipo turbidimétrica del 2,4 dinitrofenilhidrazina (DNPH) o reactivo de Brady, que se caracteriza por la obtención y visualización de un precipitado de color naranja-amarillo (turbidimétrica), tras la identificación de alfa cetoácidos en la muestra biológica de análisis.(6)
Aspectos relacionados con el incremento de los niveles de tirosina pueden estar causados por diferentes factores como la hipertirosinemia benigna transitoria en el recién nacido y la no presentación de niveles elevados del aminoácido en sangre antes de las 48 horas de vida, los cuales hacen que la cuantificación de los niveles de tirosina, no sea un marcador sensible ni específico para la TT1, pues pueden resultar en algunos falsos positivos y falsos negativos.(7) Teniendo en cuenta esta complejidad metabólica, la confirmación de la TT1 lleva aparejado una combinación de métodos con una tecnología altamente costosa. Técnicas como la cromatografía gaseosa acoplada a espectrometría de masas (CG/EM) para la detección de ácidos orgánicos y succinilacetona, la cromatografía líquida de alta resolución (HPLC) para la detección de tirosina y fenilalanina en suero, la espectrometría de masas en tándem (MS/MS) y la cromatografía de capa fina, este último para la identificación de los niveles de tirosina y metionina, constituye la tecnología usada para el diagnóstico de esta enfermedad.(8)
El diagnóstico temprano de este padecimiento puede ser sugerido por pruebas de tamizaje neonatal, aun cuando el recién nacido es asintomático.(7) En Cuba, solamente se pesquisan seis enfermedades: la fenilcetonuria, el déficit de biotinidasa, la galactosemia, el hipotiroidismo congénito, la fibrosis quística e hiperplasia adrenal congénita; razón por la cual la sospecha inicial de las TT1 está dada por las manifestaciones clínicas y posteriormente los hallazgos del laboratorio mediante el análisis de los marcadores bioquímicos, los cuales permitirán orientar el diagnóstico.(9)
El laboratorio de genética bioquímica del Centro Nacional de Genética Médica, tiene como misión la realización de estudios bioquímicos necesarios para el diagnóstico, evolución y evaluación del pronóstico de los errores innatos del metabolismo (EIM), entre los que se encuentra la TT1. El departamento, dotado de una tecnología altamente costosa, realiza una serie de determinaciones, entre las que cuentan pruebas químicas cualitativas para la identificación de aminoacidurias, la cromatografía en capa delgada de aminoácidos, la cromatografía líquida de alta resolución para la cuantificación de aminoácidos como la fenilalanina y la tirosina, así como la identificación de ácidos orgánicos y succinilacetona por la cromatografía gaseosa acoplada a espectrometría de masas.
El presente trabajo tiene como objetivo: implementar una metodología de trabajo para la detección de metabolitos marcadores bioquímicos de la tirosinemia tipo 1.
MÉTODOS
Se realizó un estudio descriptivo y transversal en una serie de casos evaluados en el período comprendido entre enero del 2021 a febrero del 2023, en el Laboratorio de Genética Bioquímica del Centro Nacional de Genética Médica, con motivo de indicación de sospecha de un error innato de metabolismo (EIM).
Muestras biológicas utilizadas en el estudio
Se utilizaron muestras de orina de la primera micción de la mañana, con ayuno superior a 6 horas y muestras de suero no hemolizadas.
Métodos
- Prueba química cualitativa de α nitroso β naftol (prueba colorimétrica)
Se añadió previamente 1000 µL de disolución de ácido nítrico 2,63N, 500 µL de disolución de α nitroso β naftol al 0,1 % y 50 µL disolución de nitrito de sodio al 2,5 % en el tubo de ensayo. Seguidamente se homogenizó y se añadió 150 µL de muestra de orina de primera micción de la mañana. Tras 10min de reacción se observó el resultado del estudio.
- Prueba química cualitativa de 2,4 dinitrofenilhidrazina (prueba turbidimétrica)
La muestra de orina de primera micción de la mañana, fue primeramente centrifugada a 500RPM por 3min, luego se extrajo el sobrenadante y se trabajó con 500µL de esta muestra. Se añadió 500 µL de la disolución de 2,4DNPH al 0,1 % y se homogenizó. Tras 10min de reacción se observó el resultado del estudio.
Cuantificación simultánea de fenilalanina y tirosina en suero
Se trabajó con 150 µL de muestra de suero, a la cual le fue añadido 150 µL de disolución de ácido tricloroacético al 5 % y se homogenizó. Posteriormente se centrifugó a 16000 rfc durante 10 minutos, se extrajo el sobrenadante y se trabajó con 100 µL de la muestra. Posteriormente fueron añadidos 400 µL de agua destilada, homogenizándose la mezcla.
Condiciones cromatográficas para el análisis por cromatografía líquida de alta resolución (HPLC)
Se empleó una técnica isocrática en fase reversa con detección directa por fluorescencia (Ex: 260 nm; Em: 282 nm) validada para la cuantificación simultánea de ambos aminoácidos en suero.
Todas las determinaciones se realizaron a 30°C y se emplearon pre-columna en fase reversa (RP-18) y columna en fase reversa (RP-18) 150 mm x 4 mm, 5 µm, (Shimadzu o Merck).
Fase móvil etanol absoluto al 5 % en agua destilada. El volumen de inyección de 20 µL, con dilución previa de las muestras con agua destilada 1: 4 (sobrenadante: agua destilada).
El tiempo de corrida fue de 7 minutos. Los metabolitos fueron identificados por su tiempo de retención y fluorescencia.
Intervalo de referencia para los dos criterios empleados en el diagnóstico de la tirosinemia en lactantes
- Tirosina (primer criterio): 0,9-1,41 mg/dL (51-79 μM).
- Fen/Tir (segundo criterio): 0,48-1,71.
- Niños con tirosinemia: > 1,41 mg/dL (> 79 μM) y la relación Fenil/Tir 0,1-0,4.
Determinación de ácidos orgánicos y succinilacetona en orina
Para preparar las muestras de orina se empleó una modificación del método experimental descrito por Duez y cols.(10) para el análisis cuantitativo de ácidos orgánicos en orina. En un tubo de ensayo se añadió la cantidad de orina equivalente a 0,25 mg de creatinina y se trató con 60 U de ureasa durante 30 min a temperatura ambiente. La muestra se alcalinizó con hidróxido de sodio (8 N) hasta pH 14 y se oximó con cloruro de hidroxilamina (40 %). Posteriormente se acidificó con ácido clorhídrico (6 N) hasta pH 1 y se saturó con 1 g de cloruro de sodio. Luego se realizaron dos extracciones líquido/líquido con 6 mL de acetato de etilo. La fase orgánica se recuperó y secó en sulfato de sodio anhidro y se evaporó a 50°C en atmósfera de nitrógeno. Una vez secas las muestras, se derivatizó con 2, 2,2-trifluoro-N-metil-N-rifluoroacetamida durante 20 min a 60°C y se inyectó 1 μL en el CG/EM con una relación de split 1/10.
Condiciones cromatográficas para el análisis por cromatografía de gases y espectrometría de masas
El procesamiento analítico para el análisis de ácidos orgánicos se efectuó en un CG/EM (QP‑2010S, Shimadzu, Kioto, Japón) con una columna capilar SLB-5ms de 30 m de longitud, diámetro interno 0,25 mm, espesor de película de 0,25 μm, con helio como gas portador. Las condiciones de adquisición del equipo fueron: temperatura del inyector de 250°C, temperatura de la interface de 280°C y temperatura del horno de 80°C hasta 300°C, con una rampa de temperatura de 7°C por min. Todos los compuestos se identificaron por su espectro de masas y su tiempo de retención.
En el algoritmo bioquímico propuesto en este estudio se realizaron de manera inicial las PQC en muestras de orina de primera micción de la mañana (pueden ser utilizadas posteriormente en el estudio de CG-EM). Teniendo en cuenta las siguientes variantes:
- Prueba del α nitroso β naftol y 2,4 DNPH positiva.
- Prueba del α nitroso β naftol positiva y 2,4 DNPH negativa.
Se solicita el envío de muestra de suero para la cuantificación simultánea de los niveles de tirosina y fenilalanina. Si el estudio por HPLC reporta niveles elevados de tirosina, se realiza el examen confirmatorio por CG/EM, en el laboratorio de espectrometría de masas, si resultan positivos aquellos fluidos biológicos cuyos perfiles cromatográficos sugerirían una TT1. Es importante destacar que la muestra extraída del paciente, una vez que el facultativo tenga la sospecha de esta enfermedad, deberá ser en el momento de la descompensación metabólica. Si la muestra es escasa para el estudio, se solicita el envío de mayor cantidad de dicha muestra. (Fig. 1).
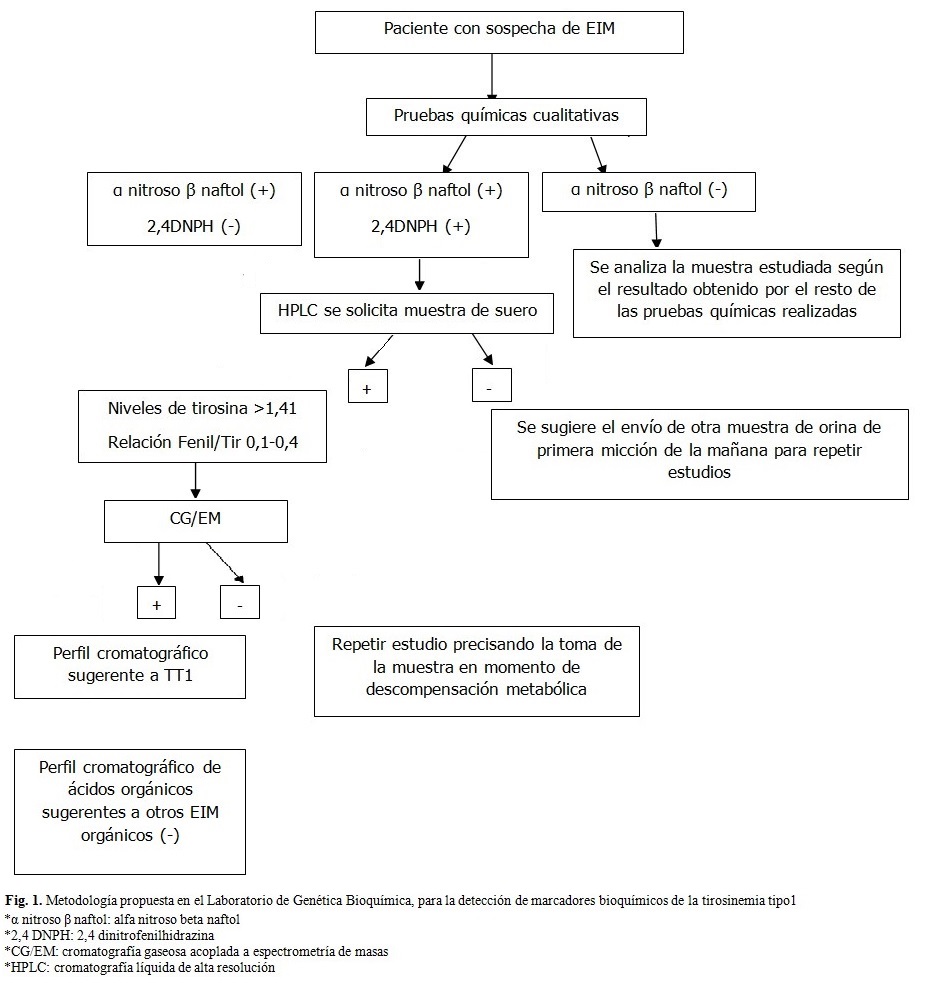
Para la recopilación de la información se utilizaron las bases de datos de entrada de muestras y emisión de resultados del Laboratorio de Genética Bioquímica del Centro Nacional de Genética Médica, específicamente de los laboratorios de pruebas metabólicas, análisis de aminoácidos y espectrometría de masas
El estudio fue aprobado por el comité de ética y de investigación del Centro. Fueron procesados muestras de pacientes de todo el país, cuyos tutores legales ofrecieron su consentimiento para utilizar los resultados de sus análisis en publicaciones científicas.
RESULTADOS
Del total de muestras analizadas, 67 resultaron positivas solamente a la prueba del α nitroso β naftol (7,4 %), 6 resultaron positivas simultáneamente a la prueba del α nitroso β naftol y del 2,4DPH que representa el 0,6 %. (Tabla 1).

Se observa el total de muestras y su positividad, después de la realización del estudio de cuantificación de la tirosina por HPLC. Fueron procesadas un total de 40 muestras de suero, resultaron positivas, 12 (30 %). (Tabla 2).

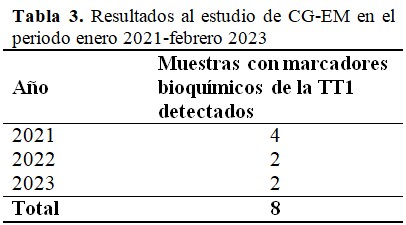
De las 12 muestras de orina de primera micción de pacientes con todas las pruebas del algoritmo bioquímico propuesto, resultaron positivas 8 (66,6 %) a metabolitos marcadores de la TT1, por el estudio de CG-EM. (Tabla 3).
DISCUSIÓN
El diagnóstico de la TT1 constituye en la actualidad un reto para los laboratorios que se especializan en el estudio de esta entidad, en especial en Cuba, donde no se realizan detecciones neonatales en papel de filtro, por lo que su diagnóstico radica en la pericia de los clínicos tras los hallazgos por la sintomatología clínica acompañada de los marcadores bioquímicos que se puedan confirmar en el los laboratorios de análisis. En la actualidad, en países como España, (que cuenta con una red de 15 laboratorios donde se realizan tamizajes neonatales por espectrometría de masas en tándem, teniendo en cuenta que, con la implementación de esta tecnología, aumenta la diversidad de enfermedades pesquisadas) se estableció el diagnóstico de 7 enfermedades que debían incluirse en todos los programas, dentro de las cuales no cuenta la TT1. Aunque existan estudios que demuestren la sensibilidad del parámetro de detección de la succinilacetona en sangre para el diagnóstico de esta enfermedad, este no se usa como principal elemento en los países que cuentan con este tipo de programa.(11,12)
De un total de 900 muestras de orina de pacientes con sospecha de EIM analizadas en el laboratorio de pruebas metabólicas (laboratorio donde se realizan los estudios cualitativos), 73 fueron positivas a las pruebas químicas cualitativas, en algunas de sus variantes. Es importante destacar, que en todas las muestras procesadas se cumplió correctamente el proceso de centrifugación, lo cual permitió la limpieza y/o ausencia de turbidez antes de realizársele la prueba turbidimétrica (2,4 DNPH). Estudios realizados reportan la positividad de estas pruebas (en alguna de sus variantes) ante la presencia de tirosinemia tipo1.(13,14) Las muestras que resultaron negativas, se les realizó el algoritmo según el resultado del resto de las PQC. Se resalta que en la metodología propuesta el punto de partida del algoritmo bioquímico fueron todas aquellas muestras recepcionadas, cuyo modelo de indicación refería como hipótesis diagnóstica una sospecha de EIM.
De las 73 muestras positivas a las pruebas cualitativas en alguna de sus variantes, solamente 40 siguieron el algoritmo establecido por el laboratorio, lo que representó el 54,8 % de los casos estudiados. La no entrada de las muestras de suero, así como la hemolisis en algunas de las enviadas, para cumplimentar la segunda etapa en el diagnóstico, fueron las causas que impidieron que se concretara el estudio en el resto de las muestras.
La aplicación de la técnica de HPLC para la cuantificación de los niveles de tirosina y fenilalanina, y de su relación, tras la positividad en 12 de sus muestras, permitió identificar el incremento de los niveles de tirosina, así como valores de la relación de Fenil/Tir por debajo de 0,41 condición para ser confirmadas por la técnica de CG-EM. A pesar de lo mostrado en estudios realizados, en algunos países, se usa el nivel de tirosina en sangre como prueba definitoria para el diagnóstico de la TT1.(15) En el laboratorio donde se realizó este estudio forma parte del algoritmo bioquímico, pero no define el diagnóstico final.
La aplicación de la técnica de CG/EM, tras la positividad en 12 de sus muestras en el estudio de cuantificación del metabolito tirosina, permitió identificar 8 pacientes con marcadores bioquímicos sugerentes a una TT1 (66,6 %). En estos, se pudo identificar aumento marcado de ácido láctico, 4-hidroxifenilpirúvico, 4-hidroxifeniláctico, 4-hidroxifenilacético.(16) Es importante destacar, que a pesar de tener establecido el método de detección de la succinilacetona por CG-EM, esta no ha podido ser identificada con valores detectables al estudio, por lo que se ha trabajado con los metabolitos antes mencionados, los cuales son reportados en la literatura como marcadores bioquímicos de la enfermedad, de conjunto con los resultados del algoritmo bioquímico y la clínica del paciente.(17,18) (Fig. 2).

La TT1 no se encuentra dentro del grupo de enfermedades metabólicas pesquisadas en Cuba. Según lo mostrado en el estudio, el diagnóstico de la enfermedad en al menos 2 casos, una vez al año. Este dato advierte, según los nacimientos al año en Cuba (aproximadamente 100000), que existan casos que no puedan resultar diagnosticados.
La negatividad obtenida en 4 de las 12 muestras estudiadas, estuvieron dadas porque la hipertirosinemia reportada por los estudios de HPLC, pudieron estar causados por factores como las tirosinemias benignas transitorias en el recién nacido, razón por la cual este estudio no se reporta como un marcador sensible en el diagnóstico.(7)
Es importante destacar en el laboratorio, ya se encuentra re-estandarizado el análisis por cromatografía en placa delgada de aminoácidos en suero y orina (TLC), como otra técnica a utilizar en la detección de niveles plasmáticos incrementados de tirosina y metionina(19) la cual se incorporará en el protocolo propuesto, para conformar la segunda etapa después de los estudios cualitativos. La poca disponibilidad de algunos patrones de metabolitos característicos del perfil metabólico de esta enfermedad, constituiría una limitante para futuros estudios, razón por la cual los facultativos que tienen la misión de sospechar e indicar la realización de este tipo de ensayos, deben de ser lo más certeros posibles para garantizar así un mejor aprovechamiento de los recursos. Cabe destacar que, en trabajos revisados en la literatura, como en los laboratorios del Instituto de Nutrición y Tecnología de los Alimentos (INTA) en Chile, el Laboratorio de Errores Innatos del Metabolismo de Costa Rica, establecen algoritmos clínicos bioquímicos similares a los desarrollados por el Centro Nacional de Genética Médica y por consiguiente una buena respuesta en cuanto a la correlación de los estudios.(16,20)
La implementación de la metodología resultó ser una herramienta valiosa en el diagnóstico temprano de la enfermedad.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Iovana Fuentes Cortés, Beliany Pacheco Suárez.
2. Curación de datos: Iovana Fuentes Cortés, Beliany Pacheco Suárez.
3. Análisis formal: Iovana Fuentes Cortés, Beliany Pacheco Suárez, Dulce María Charón Savón.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: Iovana Fuentes Cortés, Beliany Pacheco Suárez, Dulce María Charón Savón.
6. Metodología: Iovana Fuentes Cortés, Beliany Pacheco Suárez, Dulce María Charón Savón.
7. Administración del proyecto: Iovana Fuentes Cortés.
8. Recursos: Dulce María Charón Savón.
9. Software: Dulce María Charón Savón.
10. Supervisión: Iovana Fuentes Cortés.
11. Validación: Iovana Fuentes Cortés.
12. Visualización: Beliany Pacheco Suárez.
13. Redacción del borrador original: Iovana Fuentes Cortés, Beliany Pacheco Suárez, Dulce María Charón Savón.
14. Redacción – revisión y edición: Iovana Fuentes Cortés, Beliany Pacheco Suárez, Dulce María Charón Savón.
REFERENCIAS BIBLIOGRÁFICAS
- National Center for Biotechnology Information. OMIM: Online Mendelian Inheritance in Man [Internet]. Bethesda: NLM; 2020 [citado 11 Abr 2023]. Disponible en: https://www.ncbi.nlm.nih.gov/omim [Buscar en Google Scholar]
- Van Vliet K, Van Ginkel W, Jahja H, MacDonald A, Santra S, Laet C, et al. Neurocognitive outcome and mental health in children with tyrosinemia type 1 and phenylketonuria: A comparison between two genetic disorders affecting the same metabolic pathway. J Inherit Metab Dis. 2022;45(5):952-62 [Buscar en Google Scholar]
- Ginkel WG, Rodenburg IL, Harding CO, Hollak CE, Heiner MB, Van Spronsen FJ, et al. Long-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1. Paediatric Drugs. 2019;21(6):413-26 [Buscar en Google Scholar]
- Stinton C, Geppert J, Freeman K, Clarke A, Johnson S, Fraser H, et al. Newborn screening for Tyrosinemia type 1 using succinylacetone - a systematic review of test accuracy. Orphanet J Rare Dis. 2017;12(1):48 [Buscar en Google Scholar]
- Mitchell GA, Lambert M, Tanguay RM. Hypertyrosinemia. En: Scriver CR, Beaudet AL, Sly WS, Valle D. The metabolic and molecular bases of inherited disease [Internet]. Massachusetts: McGraw-Hill; 1995 [citado 20 Abr 2023]. Disponible en: https://ommbid.mhmedical.com/book.aspx?bookID=2709.p.1077-105 [Buscar en Google Scholar]
- Mesa NR, Carmona CA, Burgos LC. Pruebas bioquímicas para la detección de metabolitos producidos en los errores innatos del metabolismo. IATREIA [Internet]. 2014 [citado 15 Abr 2023];27(4):[aprox. 10p]. Disponible en: https://www.scielo.org.co/scielo.php?script=sci_arttext&pid=S0121-07932014000400005 [Buscar en Google Scholar]
- Gómez M, Danglot C, Vega L. Pautas para el diagnóstico temprano de los errores innatos del metabolismo. Rev Mex Pediatr [Internet]. 2006 [citado 16 Abr 2023];73(3):[aprox. 8p]. Disponible en: https://www.medigraphic.com/cgi-bin/new/resumen.cgi?IDARTICULO=8684 [Buscar en Google Scholar]
- Chinsky JM, Singh R, Ficicioglu C, van Karnebeek CD, Grompe M, Mitchell G, et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19(2):101 [Buscar en Google Scholar]
- Cantú C, Zepeda LM, R, Benavides S, González AS, Vázquez S, Cruz H. Incidence of Inborn Errors of Metabolism by Expanded Newborn Screening in a Mexican Hospital. Inb Errors Metab J. 2016;4(1-8):9027 [Buscar en Google Scholar]
- Cabrera N, Toledo AM. Los estudios de pesquisa activa en Cuba. Rev Cubana Salud Pública [Internet]. 2008 [citado 11 Abr 2023];34(1):[aprox. 10p]. Disponible en: https://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864-34662008000100015 [Buscar en Google Scholar]
- Duez P, Kumps A, Mardens Y. GC-MS profiling of urinary organic acids evaluated as a quantitative method. Clin Chem. 1996;42(10):1609-15 [Buscar en Google Scholar]
- Martín A, Palomino L, Ruiz P, Navarrete R, Cambra A, Quijada P, et al. Diagnosis of inborn errors of metabolism within the expanded newborn screening in the Madrid region. JIMD Rep. 2022;63(2):146-161 [Buscar en Google Scholar]
- Toapanta AE. Tirosinemia tipo I [Internet]. Chimborazo: Escuela Superior Politécnica de Chimborazo; 2018 [citado 20 Abr 2023]. Disponible en: https://dspace.espoch.edu.ec/handle/123456789/9050 [Buscar en Google Scholar]
- Das AM, Mayorandan S, Janzen N. Diagnosis Hepatorenal Tyrosinaemia in Europe: Newborn Mass Screening Versus Selective Screening. En: Tanguay R. Hereditary Tyrosinemia. Pathogenesis, Screening and Management [Internet]. 1st. ed. Canadá: Springer; 2017 [citado 18 Abr 2023]. Disponible en: https://link.springer.com/book/10.1007/978-3-319-55780-9 [Buscar en Google Scholar]
- Navarrete R, Leal F, López AM, García MT, Martín E, Quijada P, et al. Value of genetic analysis for confirming inborn errors of metabolism detected through the Spanish neonatal screening program. Eur J Hum Genet. 2019;27(4):556-62 [Buscar en Google Scholar]
- Barrera LA. Estudios bioquímicos de los errores innatos del metabolismo en Colombia, durante dos décadas. Rev Acad Colomb Cienc Exac Fis Nat [Internet]. 2009 [citado 20 Abr 2023];33(128):[aprox. 15p]. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=4086375 [Buscar en Google Scholar]
- Pérez C, Del Toro M, Díaz MC, Jara P. Protocolo de diagnóstico y tratamiento de Tirosinemia tipo I o hepato-renal. Centro de Diagnóstico de Enfermedades Moleculares [Internet]. Madrid: Universidad Autónoma de Madrid; 2022 [citado 23 Oct 2023]. Disponible en: https://aecom.com.es/protocolos/protocolo8.pdf [Buscar en Google Scholar]
- Alvear CC, Ubarnes B, Pretel C. Errores innatos del metabolismo: experiencia de trece años de estudio en Cartagena de Indias, Colombia. Rev Cien Bioméd [Internet]. 2021 [citado 15 Abr 2023];10(2):[aprox. 12p]. Disponible en: https://www.imbiomed.com.mx/articulo.php?id=115497 [Buscar en Google Scholar]
- Raimann E. Diagnóstico de errores innatos del metabolismo. Rev Chil Pediatr [Internet]. 2008 [citado 15 Abr 2023];79(Suppl. 1):[aprox. 4p]. Disponible en: https://www.imbiomed.com.mx/articulo.php?id=115497 [Buscar en Google Scholar]
- Artuch R, Moreno RM, Puig M, Montero A. El laboratorio en el diagnóstico de las enfermedades raras. Anales Sis San Navarra [Internet]. 2008 [citado 14 Abr 2023];31(Suppl. 2):[aprox. 13p]. Disponible en: https://scielo.conicyt.cl/pdf/rcp/v79s1/art14.pdf [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129