
Presentaciones de casos
Síndrome de Klippel-Trenaunay-Weber. Presentación de un caso y revisión de la literatura
Klippel-Trenaunay-Weber Syndrome. Case Report and Literature Review

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2023-02-01 08:35:52
Aprobado: 2023-06-14 13:50:17
Correspondencia: Laura Vázquez Pis. Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos. juancarlos@hosped.cfg.sld.cu
RESUMEN
Palabras clave: enfermedades raras; pediatría; informe de casos
ABSTRACT
Key words: rare diseases; pediatric; case reports
INTRODUCCIÓN
Las anomalías vasculares congénitas son difíciles de clasificar. De acuerdo a la Sociedad Internacional para el Estudio de Anomalías Vasculares, el síndrome de Klippel-Trenaunay-Weber, se caracteriza por manchas del vino de Oporto, venas varicosas e hipertrofia ósea o de tejidos blandos y otras anomalías vasculares como fistulas arteriovenosas, la presencia de solo dos de los elementos antes mencionadas se puede hacer diagnóstico de este síndrome.(1)
El síndrome de Klippel-Trenaunay-Weber (SKTW) fue descrito inicialmente en 1900 por los investigadores Maurice Klippel y Paul Trénaunay bajo el nombre de nevus varicoso osteohipertrófico. Posteriormente en 1918, Parkes Weber describió otros tres pacientes con los mismos hallazgos clínicos asociados a fistulas arteriovenosas profundas por lo que se reservó para estos casos el nombre de síndrome de Klippel-Trenaunay Weber.(1)
La patogenia de esta entidad no está clara. Existen varias teorías para explicarla: la alteración en el mesodermo fetal, se cree que se produce por un desorden del desarrollo embrionario de los tejidos mesodérmicos en el feto, que afecta a las líneas angioblástica, linfoblástica y osteoblástica, o que se deba a un daño intrauterino que origina dilatación de las anastomosis arteriovenosas. Otra teoría postula que la obstrucción y/o atresia venosa profunda causa hipertensión venosa crónica, y como resultado, aparecen en manchas vino do Oporto, varices e hipertrofia de los miembros.(2)
El gen VG5Q lo codifica y controla el crecimiento de los vasos sanguíneos, la supresión de este gen inhibe la formación de los vasos sanguíneos, definiéndose dos defectos genéticos de VG5Q el primero es una mutación de translocación incrementando su transcripción, la segunda es en el alelo E 133k encontrado en 5 pacientes con diagnóstico del SKTW.(1,2,3)
El síndrome de Klippel-Trenaunay -Weber tiene una baja incidencia de 1 en 100,000 recién nacidos, ocurre en forma esporádica, a pesar de que algunos casos familiares se hayan relatado, sin predilección por sexo ni etnias.(3)
La enfermedad tiene una expresión clinica variable, con gran diversidad de fenotipos con malformaciones asociadas. La severidad puede ser clasificada en función del tipo de displasia de los vasos sanguíneos implicados.(4,5)
El síndrome generalmente se identifica después del nacimiento. Es importante obtener un diagnóstico rápido y preciso, así como una atención médica adecuada, para tratar los síntomas y evitar complicaciones. Aunque no existe una cura para este síndrome, el objetivo del tratamiento es aliviar los síntomas y prevenir complicaciones.
Las complicaciones del síndrome de Klippel-Trenaunay-Weber pueden deberse al desarrollo anormal de los vasos sanguíneos, los tejidos blandos, los huesos y el sistema linfático. Estas pueden incluir lo siguiente: complicaciones de la mancha en vino de Oporto, malformaciones venosas, crecimiento excesivo de huesos y de tejido blando, malformaciones del sistema linfático y dolor crónico.
Se presenta el caso por lo poco frecuente de este padecimiento para mostrar su seguimiento y además con fines docentes.
PRESENTACIÓN DEL CASO
Se presenta el caso de una paciente de 5 años de edad, sin antecedentes patológicos familiares, que presentó desde el nacimiento una extensa mancha color rojo-vino localizada en el miembro inferior izquierdo. Inicialmente se le diagnosticó como hemangioma vascular, sin otros datos de interés.
Durante el primer año de vida, con el inicio de la deambulación se constató asimetría y aumento de volumen del miembro inferior izquierdo. Luego durante la primera infancia la paciente presentó molestias en el miembro inferior izquierdo a nivel de pie y tobillo sobre todo al caminar, con dificultad para realizar actividades propias de esa edad como saltar y con el antecedente de presentar cuadros de celulitis en el miembro en varias ocasiones.
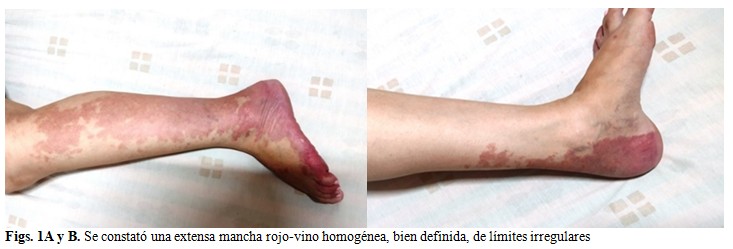
La paciente acudió al Hospital Pediátrico Universitario Paquito González Cueto, en la exploración física se constató una extensa mancha rojo-vino homogénea, bien definida, de límites irregulares, que no desaparecía a la dígito-presión, no dolorosa a la palpación, que recorría la extremidad inferior izquierda con localización en la región antero-posterior con thrill palpable en todo el trayecto venoso, compatible con un hemangioma con pulsos perifericos presentes, buen llenado capilar y movilidad conservada de la extremidad, asociada a la presencia de asimetría de ambos miembros inferiores con aumento de largo del miembro inferior izquierdo y aumento de volumen de los tejidos blandos de dicha extremidad de consistencia blanda, suave a la palpación y no era dolorosa. (Figs. 1A y B).
Exámenes complementarios
Los exámenes hematológicos (eritrosedimentacion, conteo de reticulocitos, conteo de plaquetas, coagulograma), química sanguínea se encontraron dentro de rangos normales.
Estudios por imágenes
El ultrasonido (UTS) abdominal mostró órganos del hemiabdomen superior normales, sin hepatoesplenomegalia.
La ecografía Doppler venosa de miembro pélvico izquierdo, reveló la presencia y permeabilidad del sistema venoso profundo y superficial con áreas de insuficiencia sin datos de trombosis venosa profunda.
La radiografía simple mostró la diferencia en el tamaño de ambos miembros pélvicos atribuible a las partes blandas y óseas del lado izquierdo, con un incremento de la longitud de 0,8 cm. de la extremidad izquierda y una diferencia de altura de las crestas ilíacas de 1,5 cm.
En la resonancia magnética del miembro inferior izquierdo, se identificó aumento de la trama vascular, vasos linfáticos y presencia de várices.
La arteriografía periférica de miembro pélvico izquierdo mostró flujo retrogrado del sistema venoso, y se constató, presencia de fistulas arteriovenosas.
La paciente fue evaluada por los Servicios de Angiología, Pediatría, Dermatología y Genética, y en conjunto, se llegó al diagnóstico de un síndrome de Klippel-Trenaunay-Weber. Este diagnóstico se confirmó por los datos obtenidos al interrogatorio, el examen clínico y los datos recogidos de las etapas tempranas de la vida, confirmados con estudios imagenológicos.
Se recomendó el uso de ropas adecuadas, según las anomalías músculo-esqueléticas que presentó, y vendaje de compresión, el uso de una media en el miembro afectado para prevenir la aparición de linfedema e insuficiencia venosa crónica, el uso de calzado ortopédico para la corrección de la diferencia de longitud del miembro. Como tratamiento farmacológico se prescribió el propanolol.
Actualmente se mantiene seguimiento por consulta especializada de angiología, ortopedia, pediatría, dermatología, psicología y terapia física y rehabilitación. Se mantienen con tratamiento ambulatorio sin presentar complicaciones.
DISCUSIÓN
Según las recomendaciones de la Sociedad Internacional para el estudio de Anomalías Vasculares, el síndrome de Klippel-Trenaunay-Weber se incluye dentro del grupo de las anomalías vasculares combinadas.(5)
Aunque se desconoce su verdadera etiopatogenia, se postula un defecto morfogenético que modifica la angiovasculogénesis por un mecanismo de interferencia apoptótica durante la vida intrauterina y la participación alterada de un factor antagonista vascular: la angiopoyetina II.(3)
En individuos heterocigóticos portadores de una mutación, puede ser transmitida por muchas generaciones de manera desapercibida, esta se hace evidente cuando pierde la heterocigocidad. Aunque algunos autores sugieren una posible herencia autosómica dominante, quizá una mutación somática de un gen letal que sobrevive por mosaicismo, sea la explicación más factible.(3) Las malformaciones vasculares y capilares constituyen las anomalías más frecuentes, encontrándose entre el 80 y el 98 % de los casos y las várices entre el 70 y el 80 %, siendo la hipertrofia de la extremidad el hallazgo más variable entre el 50 y el 94 %. La localización puede ser en casi todas las partes del cuerpo, pero es más frecuente en los miembros inferiores (95 %), la mayoría de los pacientes muestran afectación de un cuadrante y fundamentalmente del lado derecho(6,7,8,9) también puede afectar los brazos, tronco, es más rara su aparición en la cara, la cabeza o en los órganos internos. Las alteraciones linfáticas se observan en el 70 % de los casos, y pueden resultar en linfedema, linforrea y susceptibilidad a presentar celulitis como en este paciente.(7) En el caso reportado la afectación es en la extremidad inferior izquierda constatándose al examen físico, thill palpable en todo el trayecto venoso, sugiriendo la presencia de fístula arteriovenosas posteriormente confirmada por arteriografía periférica.
La malformación capilar, también conocida como mancha en vino de Oporto, nevo telangiectásico o angioma plano, es la manifestación cutánea más frecuente, desde el nacimiento, se asocia a una mayor frecuencia de malformaciones linfáticas y complicaciones como infecciones, sangrado, cuya intensidad de coloración puede aumentar con la edad.(7)
Algunos autores(10,11) atribuyen el sobrecrecimiento de la extremidad a un fenómeno de hipertensión venosa por trombosis o atresia del sistema venoso profundo o disregulación en la integración de canales vasculares. Puede ser secundaria a un aumento del largo o de la circunferencia que se presenta desde el nacimiento, puede ser desfigurante con el tiempo y puede ocasionar discapacidad.(7) En el caso que se presenta hubo incremento de la longitud de la extremidad izquierda y una diferencia de altura de las crestas ilíacas.
Las complicaciones se muestran como cuadros de celulitis recurrente que mejora con antibióticos. Los dolores pueden llegar a ser intensos y de inicio súbito y con la aparición de tromboflebitis superficiales y limitadas, por lo que se deben usar analgésicos durante tiempos prolongados y se recomienda no ligar la vena. La trombosis venosa profunda puede presentarse hasta en un 4 % de los casos.(10) Las complicaciones pelvianas van desde la presencia de hematuria, hemorragia gastrointestinal y estreñimiento, hasta la obstrucción del meato vesical.(11) Se describen otras complicaciones como: sobreinfección, dolores neuropáticos, parestesias, úlceras, osteomielitis, coagulopatía, insuficiencia cardíaca, hemotórax y muestran una gran predisposición a las fracturas.(12)
El diagnóstico es esencialmente clínico y se corrobora con las técnicas imagenológicas principalmente en la radiografía simple, resonancia magnética, así como el estudio imagenológico no invasivo para la evaluación de pacientes con malformaciones vasculares, la ecografía Doppler, así como la flebografía contrastada en la medida de lo posible. La arteriografía y angiografía son importantes para realizar los diagnósticos diferenciales.(13,14)
La terapéutica está dirigida a mejorar los síntomas del paciente y corregir las consecuencias de lesiones graves y la discrepancia de largo. El tratamiento de esta entidad es principalmente sintomático y conservador, fundamentalmente con compresión y tratamiento del dolor, además de antibióticos, corticoides, pero también quirúrgico, con terapia láser y radioterapia.(15,16)
Actualmente se han descrito estudios con terapia farmacológica antitumoral, para retardar la proliferación de las células musculares lisas y del endotelio vascular, la bleomicina actúa como esclerosante y el tratamiento quirúrgico se reserva en casos con presencia de abundante sintomatología. Los pacientes con hipertrofia ósea, pueden necesitar aparatos ortopédicos si la diferencia de los miembros es menor de 2 cm o cirugías para corrección de discrepancia en el largo de los miembros si mayor de 2 cm.(16)
Existen protocolos de actuación que incluyen diferentes tratamientos para controlar los síntomas y evitar complicaciones.(11, 15,16, 17)
En la terapia de compresión se envuelven las extremidades afectadas con vendas o prendas elásticas para prevenir la hinchazón, las venas varicosas y las úlceras de piel. Se recomiendan cambios en los estilos de vida y el uso de zapatos ortopédicos que pueden mejorar la salud y la función física.
La fisioterapia puede ayudar a aliviar la hinchazón de los brazos o las piernas (linfedema) y la hinchazón de los vasos sanguíneos.
La epifisiodesis es un procedimiento ortopédico que puede detener el crecimiento excesivo de las extremidades inferiores y cuando la diferencia entre ambos miembros es mayor de 2 cm, debe realizarse entre los 10 y 14 años.
La embolización es un procedimiento que se realiza mediante pequeños catéteres en las venas o las arterias, bloquea el flujo sanguíneo a determinados vasos sanguíneos.
La terapia o ablación con láser es un procedimiento que puede aclarar o eliminar los hemangiomas planos de la piel.
La ozonoterapia y la escleroterapia pueden ayudar a bloquear la vena.
La colocación de un filtro en la vena cava es un procedimiento que evita que los coágulos sanguíneos se trasladen a los pulmones.
El tratamiento quirúrgico se realiza solo si es necesario debido a la aparición de complicaciones: insuficiencia venosa, fallo cardíaco, coagulopatía de consumo y motivos estéticos.(17)
El síndrome de Klippel-Trenaunay-Weber está considerado como una enfermedad rara, por lo tanto, es poco frecuente. Tiene un patrón de herencia genética no bien definida, su manejo es multidisciplinario y se realiza tratamiento clínico sintomático encaminado a evitar la discapacidad, mejorar la capacidad funcional, calidad de vida y prevenir complicaciones.
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Laura Vázquez Pis, Maday González Hernández.
2. Curación de datos: Laura Vázquez Pis, Maday González Hernández.
3. Análisis formal: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil.
4. Adquisición de fondos: Esta investigación no contó con adquisición de fondos.
5. Investigación: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil, Antonio Masot Rangel.
6. Metodología: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil, Antonio Mazot Rangel.
7. Administración del proyecto: Laura Vázquez Pis, Maday González Hernández.
8. Recursos: Aimé Marrero Gil, Antonio Mazot Rangel.
9. Software: Aimé Marrero Gil, Antonio Mazot Rangel.
10. Supervisión: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil.
11. Validación: Aimé Marrero Gil, Antonio Mazot Rangel.
12. Visualización: Aimé Marrero Gil, Antonio Mazot Rangel.
13. Redacción del borrador original: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil, Antonio Mazot Rangel.
14. Redacción – revisión y edición: Laura Vázquez Pis, Maday González Hernández, Aimé Marrero Gil, Antonio Mazot Rangel.
REFERENCIAS BIBLIOGRÁFICAS
- Espín G, Suntaxi L, Yambay C, Silva R, Espín L, Vásquez B. Síndrome Congénito de Klippel-Trenaunay-Weber. Caso Clínico. Int J Morphol[Internet]. 2020[citado 11/9/2022];38(6):[aprox. 6p.]. Disponible en: https://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0717-95022020000601842&lng=es.
- Snee IA, Mazzola CA, Sikorskyj T. Chiari I malformation with Klippel-Trenaunay syndrome: a case report and review of the literature. Childs Nerv Syst. 2021;37(7):2369-73.
- Opdenakker O, Renson T, Walle JV. Vesical hemangioma in a patient with Klippel-Trenaunay-Weber syndrome. J Pediatr. 2019;20(8):293.
- Chimbo T, Castro Y, Rizo T. A propósito de un caso: síndrome de Klippel-Trénaunay. Rev Ecuat Pediatr[Internet]. 2018[citado 5/5/2022];19(1):[aprox. 3p.]. Disponible en: https://pesquisa.bvsalud.org/portal/resource/pt/biblio-996421.
- Barajas TJ, Delgado EG, Urióstegui LC, López V, Luna U. Síndrome de Klippel Trenaunay. Rev Cubana Med Gen Integr[Internet]. 2016[citado 15/2/2023];32(3):[aprox. 10p.]. Disponible en: https://scielo.sld.cu/pdf/mgi/v32n3/mgi17316.pdf.
- Gontero R, Ortiz A, Roverano S, Paira S. Síndrome Klippel-Trenaunay: comunicación de dos casos. Rev Argent Reumatol[Internet]. 2017[citado 8/6/2022];28(1):[aprox. 5p.]. Disponible en: https://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S2362-36752017000100006.
- Cuan G, Granado A. Síndrome de Klippel y Trénaunay. Rev Cienc Méd Pinar del Río[Internet]. 2019[citado 7/11/2022];23(6):[aprox. 4p.]. Disponible en: https://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1561-31942019000600941&lng=es.
- Razek AA. Imaging findings of Klippel-Trenaunay Syndrome. J Comput Assist Tomogr. 2019;43(5):786-92.
- Vergadi E, Goniotakis I, Maraki S, Galanakis E. Extensive Cellulitis and Bacteremia Due to Streptococcus Pseudoporcinus in a Child With Klippel-Trenaunay Syndrome. Pediatr Infect Dis J. 2021;40(8):e316-8.
- Asghar F, Aqeel R, Farooque U, Haq A, Taimur M. Presentation and management of Klippel-Trenaunay syndrome: a review of available data. Cureus. 2020;12(5): e8023.
- Ivanitskaya O, Andreeva E, Odegova N. Prenatal diagnosis of Klippel–Trenaunay syndrome: series of four cases and review of the literature. Ultrasound. 2020;28(2):91-102.
- Huang FL, Chen HY, Chang TK. Medical treatment of a female patient with complicated Klippel–Trenaunay syndrome. Pediatr Neonatol. 2018;59(5):527-30.
- Sait H, Sahi PK, Kapoor S. Portal Hypertension in a Case of Klippel Trenaunay Syndrome. Indian Pediatr. 2020;57(8):754.
- Escobar Y, Ayra CM. Síndrome de Klippel-Trenaunay en un recién nacido. Medisan[Internet]. 2019[citado 24/2/2023];23(1):[aprox. 9p.]. Disponible en: https://scielo.sld.cu/scielo.php?pid=S102930192019000100121&script=sci_arttext&tlng=en.
- Berchiolli R, Marconi M. Klippel-Trenaunay syndrome: a dramatic presentation. Eur J Vas Endovasc Surg. 2018;56(2):299.
- Hudson DA, Mohan AT, Lelala NB. Intralesional Bleomycin Injection in Management of Localized Venous Problems in Children with Klippel-Trenaunay Syndrome. Plastic Reconstr Surg. 2021;147(5):918e-9e.
- Candia RF, Palacios JM, Rosas BR. RR. Síndrome de Klippel-Trenaunay-Weber. Reporte de un caso. Rev Mex Angiol[Internet]. 2008[citado 23/6/2022];36(1):[aprox. 10p.]. Disponible en: https://www.medigraphic.com/cgi-bin/new/resumenI.cgi?IDARTICULO=42742.
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129