
Presentaciones de casos
Hipogonadismo masculino. Presentación de un caso
Male Hypogonadism. A Case Report
Especialista de I Grado en Medicina General Integral, Especialista de I Grado en Medicina Interna, Máster en Urgencias Médicas, Profesor Instructor
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2012-06-05 16:37:25
Aprobado: 2012-06-25 12:07:31
Correspondencia: Lisandro Hernández Madrazo. Policlínico Universitario Enrique Barnet, Lajas, Cienfuegos amgiraldoni@jagua.cfg.sld.cu
RESUMEN
Se presenta el caso de un paciente de 26 años, que acudió a consulta de Medicina Interna en el Centro de Diagnóstico Integral La Fortaleza, Maracaibo, Estado Zulia, Venezuela, por presentar disminución del tamaño de los genitales externos, con escaso desarrollo de estos desde su niñez, y aumento de volumen de las mamas. Al examen físico se constató un tronco de configuración feminoide por panículo adiposo acumulado en bajo vientre, mama y pubis; pelvis ancha; predominio de las extremidades inferiores sobre las superiores, proporciones eunucoides; aumento de volumen de forma difusa en ambas mamas (ginecomastia); depósito de tejido graso a nivel de la cintura pélvica; y ausente o escaso vello facial, axilar y pubiano. Se observó tamaño disminuido, escasa pigmentación, y consistencia blanda, en pene y testículos. Se le realizó examen de testosterona plasmática, hormona luteinizante y hormona folículo estimulante, concluyéndose en el servicio de Endocrinología del Hospital Universitario de Maracaibo, como un hipogonadismo hipogonadotrópico de causa no demostrable. El diagnóstico clínico de un hipogonadismo en el adulto no es habitual en la práctica médica, hecho en el cual radica la relevancia del caso presentado.
Palabras clave: hipogonadismo; testosterona; informes de casos Límites: Humano; adulto; masculino
ABSTRACT
The case of a 26 years old male patient who attended the Internal Medicine consultation at the La Fortaleza Integral Diagnostic Center in Maracaibo, Zulia State, Venezuela because of decreased external genitalia size, with poor development from childhood and swelling of the breasts is presented. Physical examination showed a trunk of feminoid configuration caused by adipose tissue accumulated in the lower abdomen, breast and pubic; wide pelvis; lower limb dominance over higher limbs; enucoid proportions; volume diffusely increased in both breasts (gynecomastia); deposit of fatty tissue at the pelvic girdle, and absent or sparse facial, axillary and pubic hair. We observed decreased size, poor pigmentation, and soft consistency in penis and testicles. Exam was performed on plasma testosterone, luteinizing hormone and follicle stimulating hormone, thus concluding, by the Endocrinology Service at the Maracaibo University Hospital, to be the case of hypogonadotropic hypogonadism of improvable cause. The clinical diagnosis of hypogonadism in adults is unusual in medical practice, a fact that provides with relevance the case we present.
Key words: hypogonadism; testosterone; case reports Limits: Human; adult; male
INTRODUCCIÓN
El aparato reproductor masculino regula la diferenciación sexual, la virilización y los cambios hormonales que acompañan a la pubertad, lo que conduce finalmente a la espermatogenia y la fertilidad. Bajo el control de las hormonas hipofisarias (hormona luteinizante [LH, del inglés luteinizing hormone] y hormona folículo estimulante [FSH, del inglés follicle-stimulating hormone]), las células de Leydig de los testículos producen testosterona y las células reproductoras (germinativas) son reguladas por las células de Sertoli en su división, diferenciación y maduración hasta transformarse en espermatozoides. Durante el desarrollo embrionario, la testosterona y la dihidrotestosterona (DHT) inducen la formación del conducto de Wolff y la virilización de los genitales externos. Durante la pubertad, la testosterona favorece el crecimiento somático y el desarrollo de las características sexuales secundarias. En el adulto es necesaria esta hormona para la espermatogenia, la estimulación de la libido y el desempeño sexual normal.1,2
El hipogonadismo masculino se define como el déficit testicular tanto endocrino como exocrino, pero es conocido que la mayoría de los pacientes con déficit de la espermatogénesis, no tienen insuficiencia hormonal ostensible. Por tanto, es preferible utilizar el nombre de hipogonadismo solo cuando existe merma de la función endocrina del testículo, y considerar al déficit exocrino como un trastorno aislado de la fertilidad o la espermatogénesis. La época en que se produce la deficiencia gonadal en relación con la pubertad, determina amplias variaciones clínicas, por lo que estas alteraciones deben considerarse en dos estadios distintos, prepuberal y pospuberal. También existen diferencias según el elemento afectado; puede verse afectada la gónada (hipogonadismo primario), la hipófisis (hipogonadismo secundario) y la región hipotalámica (llamado por algunos hipogonadismo terciario).3
El hipogonadismo masculino afecta a 4 millones de norteamericanos, de los cuales solo 5 % recibe tratamiento. Su incidencia aumenta de 12 % a los 50 años a 50 % a los 80 años.4,5
La deficiencia androgénica es una situación clínica frecuente. El síndrome de Klinefelter tiene una prevalencia de 1 en 500 recién nacidos vivos, y si se suman otras causas congénitas o adquiridas de lesiones testiculares e hipotálamo-hipofisarias, se estima que alrededor de 1 en 200 hombres presentan reducción de los niveles circulantes de testosterona (T). Si, además, se considera la deficiencia androgénica asociada al envejecimiento, esta prevalencia aumenta significativamente. El hipogonadismo, en su presentación clínica clásica, es de fácil diagnóstico, pero las formas menos severas presentan dificultades para su reconocimiento. Sin embargo, es de fundamental importancia establecer con certeza la deficiencia androgénica antes de iniciar cualquier terapia de sustitución debido a los riesgos potenciales.6 El diagnóstico se sospecha clínicamente y se establece con la demostración de concentraciones bajas de testosterona en sangre.7 En estos casos, la determinación de las gonadotropinas LH y FSH permite relacionar la insuficiencia testicular con una anomalía hipotálamo hipofisaria, cuando estas hormonas hipofisarias muestran una concentración reducida (hipogonadismo hipogonadótropo o déficit gonadótropo), o con una enfermedad primaria gonadal cuando su concentración está aumentada (hipogonadismo hipergonadotropico).1,7
Se considera de interés la publicación y divulgación de este caso, debido a que el diagnóstico clínico de un hipogonadismo en edad adulta no es habitual en la práctica médica.
PRESENTACIÓN DEL CASO
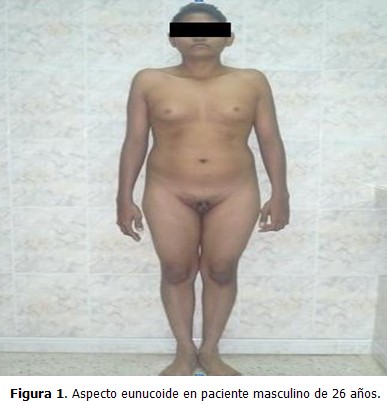
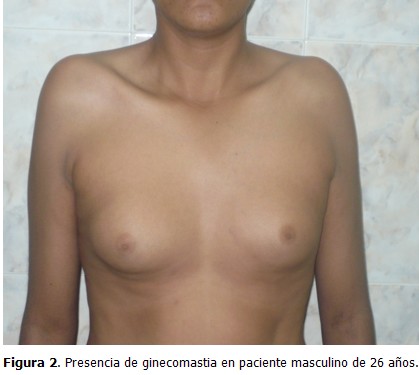
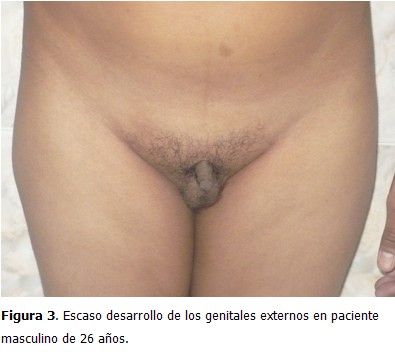
Paciente de 26 años, que acudió a consulta de medicina interna en el Centro de Diagnóstico Integral (CDI), La Fortaleza, Maracaibo, Estado Zulia, Venezuela, por presentar disminución del tamaño de los genitales externos, con escaso desarrollo de estos desde su niñez, así como aumento de volumen de las mamas, sin secreción por el pezón, dolor, o cambios de coloración en la piel. Al examen físico se constató un tronco de configuración feminoide por panículo adiposo acumulado en bajo vientre, mamas y pubis; pelvis ancha; predominio de las extremidades inferiores sobre las superiores; proporciones eunucoides (se define como envergadura de 2 cm o más que la estatura) (Figura 1); aumento de volumen de forma difusa en ambas mamas (ginecomastia) (Figura 2); depósito de tejido graso a nivel de la cintura pélvica; y vello facial, axilar y pubiano ausente o escaso. Se observó tamaño disminuido, escasa pigmentación, y consistencia blanda, en pene y testículos (Figura 3).
Se realizó tomografía de cráneo y silla turca: la primera mostró engrosamiento mucoso de seno maxilar izquierdo y celdas etmoidales, así como tabique nasal desplazado a la izquierda; mientras que la segunda mostró parámetros normales.
El ecograma testicular y prostático, reveló la presencia de testículos pequeños y próstata disminuida de tamaño, textura homogénea y contornos regulares.
Se indicó la dosificación hormonal de testosterona plasmática, hormona luteinizante (LH), hormona folículo estimulante (FSH), y se remitió al paciente al servicio de Endocrinología, concluyéndose como un hipogonadismo hipogonadotrópico de causa no demostrable. Se sumó entonces tratamiento sustitutivo con andrógenos.
No fue posible dar seguimiento a posteriori, ya que el paciente fue remitido a un nivel terciario de atención.
DISCUSIÓN
El hipogonadismo puede manifestarse con deficiencia de testosterona, infertilidad o ambas. Los síntomas de hipogonadismo dependen de la edad de aparición. Antes de la pubertad las manifestaciones son más floridas: testículos, pene y próstata pequeños, vello axilar y púbico escasos, brazos y piernas desproporcionadamente largos (por el retraso del cierre epifisario), musculatura reducida, ginecomastia y persistencia del timbre agudo de voz. La pérdida pospuberal de la función testicular produce signos y síntomas clínicos sutiles que aparecen lentamente. En general, podemos encontrar un descenso progresivo de la masa muscular, pérdida de la libido, impotencia, oligospermia o azoospermia, dificultad para la concentración y, ocasionalmente, sofocos de tipo menopausia (cuando el comienzo del hipogonadismo es agudo). El riesgo de osteoporosis y fracturas se incrementa, y muchos casos de hipogonadismos se descubren durante la investigación sobre infertilidad.8 Debido a que la hormona luteinizante y la hormona folículo estimulante son hormonas tróficas para los testículos, las alteraciones de la secreción de estas gonatropinas hipofisarias producen hipogonadismo secundario, caracterizado por concentraciones reducidas de testosterona en pacientes con valores bajos de LH y FSH. En las personas con deficiencias más graves se observa una ausencia completa de desarrollo puberal, infantilismo sexual, como en este paciente, y en algunos casos, hipospadias y criptorquidia. Los pacientes con deficiencia parcial de gonadotropinas tienen un retraso o detenimiento del desarrollo sexual. El hipogonadismo hipogonadotrópico se puede clasificar en trastornos congénitos y adquiridos. Los trastornos congénitos con frecuencia implican deficiencia de GnRH, con la consiguiente deficiencia de gonadotropinas, entre ellos se citan, Síndrome de Kallmann, Síndrome Prader -Willi, Sindrome Laurecen-Moon-Bieldl.1,2 El síndrome de Kallmann se caracteriza por deficiencia aislada de gonadotropinas y anosmia o hiposmia, debido a un desarrollo defectuoso del bulbo olfatorio. Se han descrito paladar y labio hendido, sordera, acortamiento del cuarto metacarpiano, anomalías cardíacas y epilepsia. El modo de transmisión es autosómico dominante. La incidencia del síndrome es de 1 en 10 000 nacimientos masculinos. Se presenta clínicamente con los siguientes signos: hábito eunucoide, ginecomastia, atrofia testicular, masa prepuberal y ausencia de vello corporal. Las gonadotropinas séricas están disminuidas y se normalizan administrando Gnrh pulsátil, ya que el defecto es a nivel del hipotálamo. El síndrome de Kalmann se asocia con una herencia autosómica dominante, autosómica recesiva, o ligada al cromosoma X. Esta última se debe a mutaciones y delecciones del gen KAL que está localizado en Xp22.3. Este gen está compuesto por 14 exones y codifica la síntesis de anosmia-1, una proteína asociada con funciones de adherencia celular y actividad antiproteasa.9 También hay mutaciones del gen del receptor de crecimiento de los fibroblastos (FGFR-1), lo cual conduce a agenesia de las neuronas olfatorias y secretoras de GnRH.9,10 Los trastornos adquiridos son mucho más comunes que los congénitos, y se deben a diversas lesiones ocupativas en la silla turca o a enfermedades infiltrativas del hipotálamo o la hipófisis. Los trastornos específicos incluyen adenomas hipofisarios, craneofaringioma, infarto de la hipófisis, aneurismas, enfermedad granulomatosa como la tuberculosis, sarcoidosis, hemocromatosis.1,2 Los granulomas hipofisarios de la sarcoidosis, la tuberculosis y la infiltración de la hipófisis con hierro que ocurre en la hemocromatosis, son causas relativamente raras de deficiencia de gonadotropinas. Tanto la testosterona como las gonadotropinas están disminuidas.11 El tratamiento de las lesiones cerebrales con radioterapia puede producir hipopitituarismo; ocurre lentamente y a medida que pasan los años la incidencia aumenta.7 En este paciente no se identificó la causa del hipogonadismo.
REFERENCIAS BIBLIOGRÁFICAS
- Shalender Bhasin J, Larry Jameson. Trastornos de los testículos y del aparato reproductor masculino. En: Fauci AS, Braunwald E, Kasper DL, Hauser SL, Longo DL, Jameson L, et al, editores. Harrison Principios de Medicina Interna. 17 ed. Mexico: McGraw-Hill-Interamericana; 2008: p. 2310-24 [Buscar en Google Scholar]
- Mastsumoto AM. Testículo. En: Bennet JC, Plum F, editores. Cecil.Tratado de Medicina Interna. 20 ed. México: MGraw- Hill-Interamericana; 1996: p. 1529-45 [Buscar en Google Scholar]
- Roca Goderich, Smith Smith VV, Paz Presilla E, Losada Gómez J, Senet Rodriquez B, Llanos Sierra N, et al. Enfermedades de las Gonadas. En: Temas de Medicina Interna. 4ta ed. Ciudad de la Habana: Editorial Ciencias Médicas; 2002: p. 270-83 [Buscar en Google Scholar]
- Seftel AD. Male hypogonadism. Part I: Epidemioloy of hypogonadism. Int J Impot Res. 2006;18(2):115-20 [Buscar en Google Scholar]
- Harman SM, Metter EJ, Tobin JD, Pearson J, Blackman MR. Longitudinal effects of aging on serum total and free testosterone levels in healthy men. Baltimore Longitudinal Study of aging. J Clin Endocrinol Metab. 2001;86(2):724-31 [Buscar en Google Scholar]
- Knoblovits P, Levalle O, Nagelberg A, Pacenza N, Rodríguez M. Hipogonadismo masculino. Rev argent endocrinol metab [revista en Internet]. 2007 [citado 12 Feb 2012];44(3):[aprox. 16p]. Disponible en: http://www.scielo.org.ar/scielo.php?script=sci_arttext&pid=S1851-30342007000300002 [Buscar en Google Scholar]
- Jubiz W, Cruz EA. Hipogonadismo masculino: Causas, genética, diagnóstico y tratamiento. Colomb Méd. 2007;38(1):84-91 [Buscar en Google Scholar]
- Becerra Fernández A. Sexualidad y reproducción, un campo importante en la endocrinología. Endocrinología y Nutrición. 2006;53(1):34-41 [Buscar en Google Scholar]
- Karges B, de Roux N. Molecular genetics of isolated Hypogonadotropic hipogonadism and Kallmann syndromes. Endocr Dev. 2005;8:67-80 [Buscar en Google Scholar]
- Dode C, Levilliers J, Dupont JM, De Paepe A, Le Du N, Sousssi- Yanicostas N, et al. Loss – of-functions mutations in FGFR-1 cause autosomal dominant Kalmanns syndrome. Nat Genet. 2003;33(4):463-5 [Buscar en Google Scholar]
- Jubiz W. Testículos. En: Endocrinología Clínica. 4ta ed. Cali : Editorial Feriva; 2002: p. 355-74 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129