
Presentaciones de casos
Síndrome de Marfán: a propósito de dos casos
Marfan Syndrome: Regarding Two Cases
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2020-02-26 19:37:55
Aprobado: 2020-02-26 19:39:41
Correspondencia: Elsy Roxana Geroy Moya. Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos. roxanagm@hosped.cfg.sld.cu
RESUMEN
Palabras clave: Palabras clave: síndrome de Marfán; enfermedades genéticas congénitas; enfermedades raras; diagnóstico; informes de casos
ABSTRACT
Key words: : marfan syndrome; genetic diseases inborn; rare diseases; diagnosis; case reports
INTRODUCCIÓN
El síndrome de Marfán fue informado por primera vez alrededor de unos 100 años atrás por el médico pediatra Antoine-Bernard de Marfán en 1896, quien se percató de la asociación de dígitos largos y delgados y otras anormalidades esqueléticas en una paciente de 5 años.(1)
En aquel entonces se le nombraba aracnodactilia, por la forma característica de las manos de quienes lo padecían. En 1931 Henricus Jacobus Marie Weve demostró que el trastorno era hereditario y fue el primero en usar el término síndrome de Marfán. En 1955 Víctor McKusick, médico estadounidense y pionero de la genética médica, describió el síndrome de Marfán en su monografía titulada: Trastornos hereditarios del tejido conectivo. En 1991 se descubrió que el defecto básico en el síndrome se ha atribuido a un gen fibrilina-1 alterado (FBN1) en el cromosoma 15q21.1, constituyendo la fibrilina, una proteína esencial para la formación de fibras elásticas del tejido conectivo.(2,3)
A lo largo de la historia se fueron asociando signos y sintomatologías de este síndrome hasta establecer los criterios de diagnóstico por De Paepe y cols. en1996.(1)
Este padecimiento aparece descrito dentro de algunos síndromes con talla alta disarmónica, por la presencia de signos dismórficos o de asimetrías entre los segmentos corporales asociados a macrosomía. De estos se hace el diagnóstico diferencial pues, pueden manifestarse con macrosomía al nacer, dentro de estos los hiperinsulinismos (ya sean persistentes o los transitorios de los hijos de madre diabética), el síndrome de Becwith-Wiedemann (por mutación que conlleva hiperexpresión del factor de crecimiento análogo a la insulina (IGF) o el síndrome de Sotos (macrosomía asociada a gigantismo cerebral) ya que ellos cuyo hipercrecimiento aparece ya a partir de los 2-3 años de edad, como el síndrome de Marfán, el de Klinefelter (47XXY), el de la superhembra (47,XXX), la polisomía 47XYY, entre otros.(3)
Algunos estudios señalan que este síndrome es un trastorno genético autosómico dominante y que basta heredar una sola copia del gen con la mutación para que el síndrome se manifieste. Sin embargo, existe la posibilidad de padecer del síndrome por una mutación espontánea (caso novo), como sucede en el 25% de los casos estudiados. Se ha demostrado que este gen tiene una amplia variabilidad genética, se han registrado 1847 mutaciones tanto benignas como patológicas y 1096 variantes proteicas hasta el momento; han sido algunas de ellas únicas por grupo familiar.(1,2,4,5)
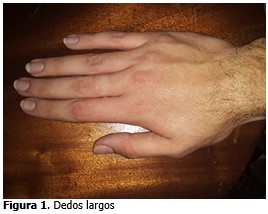
Su prevalencia es de 1 en 5 000- 10 000 recién nacidos, afecta por igual a ambos sexos. Se trata de una enfermedad multisistémica, cuyas principales manifestaciones clínicas ocurren a nivel esquelético, ocular y cardíaco. Habitualmente, la afectación esquelética es el primer signo de la enfermedad y puede incluir una dolicostenomelia (longitud excesiva de las extremidades), talla grande, aracnodactilia, hipermovilidad articular, escoliosis, protrusión del acetábulo, deformidad torácica con pectuscarinatum o excavatum, dolicocefalia, hiperlaxitud articular o hipoplasia malar. La afectación oftalmológica lleva a una ectopia o luxación del cristalino y/o una miopía axial. Asimismo, pueden presentarse signos cutáneos (estrías atróficas), riesgo de neumotórax y ectasia dural. Para el diagnóstico de síndrome de Marfán en pacientes sin antecedentes familiares de enfermedad deben estar involucrados dos órganos/sistemas que reúnan criterios mayores y al menos la afectación de un tercer órgano/sistema. En pacientes con historia familiar de síndrome de Marfán solo se requiere un criterio mayor, con datos que sugieran afectaciónde un segundo sistema.(6,7)
La alteración cardiovascular constituye la de mayor causa de morbimortalidad de esta patología, y se presenta en el 25% de los pacientes durante la edad pediátrica, e incluye dilatación de la raíz aórtica con riesgo de disección aórtica, poco frecuente en la infancia, pero de gran importancia en la adolescencia.Otras alteraciones son: el prolapso de válvula mitral y las arritmias. Estas alteraciones en el síndrome de Marfán predisponen a muerte súbita por alteraciones estructurales y funcionales.(8,9)
En el caso del prolapso de válvula mitral, es clásica su aparición en pacientes con síndrome de Marfán, aunque también está asociada a otros trastornos del tejido conectivo, como la osteogénesis imperfecta, síndrome de Ehler-Danlos y pseudoxantoma elasticum.(10,11)
Existen tres formas de presentación relacionadas con la edad, manifestaciones clínicas y pronóstico: neonatal, infantil y clásica, esta última es la más frecuente en niños, adolescentes y adultos.(7)
Su diagnóstico no siempre es simple, sobre todo en niños y adolescentes. Han sido definidos varios criterios de diagnóstico: Berlín (1986), Ghent (1996) y Ghent revisados (2010) con el objetivo de facilitar el diagnóstico clínico. En 2010, tras la revisión de los criterios de Ghent se dio mayor énfasis a las alteraciones cardíacas (dilatación/disección de la raíz aórtica), ocular (ectopia lentis) y el estudio molecular. Propuestas para agilizar y facilitar el diagnóstico clínico, así como, mejorar el diagnóstico diferencial con otras entidades relacionadas con el síndrome de Marfán.(8)
El estudio de familiares es crucial tanto para el diagnóstico precoz como para el asesoramiento genético, por lo que deben atenderles equipos multidisciplinares específicos. El trabajo coordinado con genetistas y cardiólogos, así como oftalmólogos, traumatólogos y rehabilitadores, es esencial para una atención integral a estas familias.(8,12)
La expectativa de vida en el síndrome de Marfán, condicionada fundamentalmente por el pronóstico cardiovascular, ha pasado de los 45 años en 1972, a los 72 años en 1995. Considerando que, a medida que pasa el tiempo, ha aumentado la expectativa de vida para estos pacientes, es posible inferir que se puede alcanzar también un mejoramiento de sus condiciones. Para ello, se hace necesario indagar acerca de la prescripción del ejercicio en pacientes con este síndrome, teniendo en cuenta las implicaciones, principalmente cardiopulmonares y osteomusculares que ellos presentan, con el fin de identificar cómo puede mejorarse su condición y lo que contribuirá a que haya un adecuado abordaje fisioterapéutico.(13,14)
El manejo y seguimiento se realiza por un equipo multidisciplinario. En la prevención primaria se debe realizar consejo genético y se considera la planificación familiar. Para la prevención de complicaciones: el genetista debe realizar un seguimiento por larga data, mientras que el pediatra analizará continuamente los criterios de desarrollo y crecimiento, se debe tener en cuenta la educación en la actividad física de acuerdo al grado de compromiso cardíaco. Se debe realizar valoración anual en el campo ortopédico, oftalmológico y sobre todo cardiovascular para detectar a tiempo nuevas lesiones. La frecuencia de esta última depende del grado de las lesiones. Se debe considerar además el apoyo psicológico para el niño y su familia, la educación en síntomas de pesquisa de complicaciones como neumotórax, disección aórtica y el tratamiento de complicaciones cardiovasculares.(15)
PRESENTACIÓN DE CASOS
Caso 1
Se presenta el caso de un paciente de 17 años de edad, de color de piel blanca, sexo masculino, hijo de padres no consanguíneos, producto de un embarazo que cursó con infección vaginal (moniliasis) en los 3 trimestres por lo que llevó tratamiento. La madre debutó con enfermedad hipertensiva del embarazo en el último trimestre, por lo que tuvo tratamiento con metildopa. El parto fue distócico, por cesárea a las 41,6 semanas por macrosomía fetal, el peso al nacer fue de 5120gr.
El niño tenía alta talla desde la primera infancia. El paciente tuvo tratamiento rehabilitador por deformidad torácica (pectum excavatum y genus varus). Ingresó a los 13 años por presentar episodios de lipotimias dado por debilidad muscular, fatiga, sin pérdida de conciencia.
El paciente, con antecedentes patológicos familiares de padre y abuelo paterno fallecidos en edades tempranas debido a miocardiopatía dilatada más cardiesclerosis. Ambos con fenotipo de un síndrome de Marfán y con antecedentes patológicos personales de deformidad torácica (pectum excavatum y genus varus) en la primera infancia.
Durante el ingreso se realizaron los siguientes estudios:
Electroencefalograma (EEG) y tomografía axial computarizada (TAC) de cráneo que se mostraron normales. Se evaluó y se le dio seguimiento por neurología por este cuadro.
Se valoró el caso con cardiología y se le realizó ecocardiograma y se constató un prolapso de la válvula mitral, sin otras alteraciones cardiovasculares en ese momento.
Además, se evaluó por endocrinología por alta talla y se le realizaron los complementarios:
Hormona folículo estimulante: (FSH): 1,23 muI/lT3 libre 7,40pmol/l.
Hormona luteilizante: (LH): 4,14 muI/lT4 libre15,42pmol/l.
Testosterona: 6, 58ng/ml.
Survey óseo: sincondrosis persistente de vértebras sacras. Mentón prominente.
Silla turca abalonada con aumento de sus diámetros. Sin otras alteraciones.
Sin lesiones pleuropulmonares y con una cardiomegalia ligera.
Cariotipo: 46xy (12 metafase), masculino cromosómicamente normal.
Ambas rodillas sin alteraciones óseas. Cartílagos de crecimiento pequeños.
Sin alteraciones del resto de los huesos estudiados.
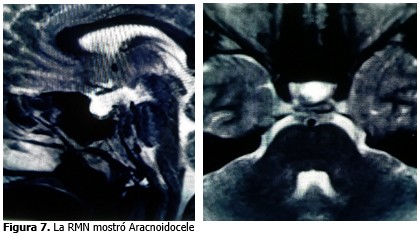
Durante el seguimiento por neurología, se indicó posteriormente resonancia magnética (RMN) con diagnóstico por este estudio de un aracnoidocelegrado III, se valoró por neurocirugía y se inició tratamiento con acetazolamida de 1 tableta cada/12 horas, prednisona (5mg) 1 tableta cada 12 horas.
Durante su evolución y seguimiento por cardiología fue llamativa la dilatación de la aorta ascendente en los últimos 3 años por lo que se evaluó en el Cardiocentro William Soler y se indicó tratamiento con losartán (50mg) ½ tableta cada 8h. Dado la progresión del cuadro fue intervenido quirúrgicamente, en diciembre del 2018.
Datos positivos al examen físico:
Piel de la espalda con presencia de estrías y hemangioma.
Talla/edad por encima del 97 percentil.
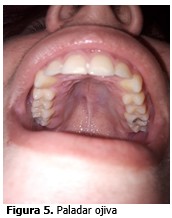
Paladar ojival.
Sistema osteomioarticular:
Dolicostenomelia (extremidades alongadas en relación a las dimensiones del tronco).
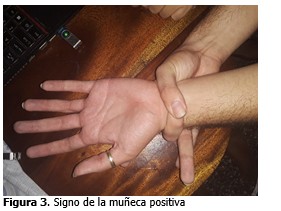
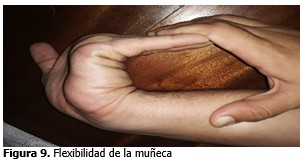
Signo de la muñeca positivo (signo de Walker Murdoch: positivo).
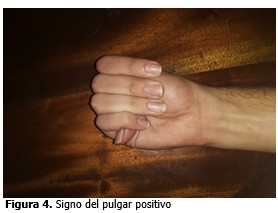
Signo del pulgar positivo (signo de Gowers: positivo).
Deformidad torácica (pectum excavatum ligero).
Laxitud articular.
Pie plano unilateral.
Examen oftalmológico con trastornos de refracción.
Tratamiento actual:
El paciente toma losartán (50mg) ½ tableta cada 8 horas para las alteraciones cardiovasculares. Tiene restricción para realizar ejercicios físicos y se le recomendó evitar la fatiga.
Se mantiene con seguimiento multidisciplinario con las especialidades de neurología, neurocirugía, cardiología, endocrinología, fisiatría, oftalmología, ortopedia y genética clínica.
La impresión diagnóstica manifestó síndrome de Marfán, aracnoidocele grado III, prolapso de válvula mitral y dilatación de la aorta ascendente corregida.
Se mantiene con seguimiento semestral, con valoración en Cardiocentro Ernesto Che Guevara de Villa Clara.
Caso 2:
Se presenta el caso de un paciente de 15 años de edad, de color de piel blanca, de sexo masculino, hijo de padres no consanguíneos, de procedencia urbana, producto de un embarazo que cursó sin alteraciones y parto a las 41,2 semanas, eutócico, con peso al nacer de 4 040 grs y Apgar adecuado. A los 6 años comenzó a presentar dolores óseos y alteraciones músculo- esquéleticas por lo cual fue evaluado en consulta de ortopedia de su área de salud, llevó seguimiento por 2 años en consulta, posterior a lo cual la madre decidió llevarlo al Hospital Frank País, donde fue evaluado por un equipo multidisciplinario que indicó la posibilidad de corrección quirúrgica de deformidades esqueléticas en miembros inferiores y seguimiento por alteraciones en columna vertebral. Además, llevaba seguimiento por nutrición por bajo peso, consulta que abandonó a los 9 años de edad.
A los 13 años regresó a consulta de nutrición nuevamente y se constató una alta talla para la edad y se solicitó interconsulta con genética clínica. Refirió el paciente en esa ocasión que venía presentando desde los 10 años de edad dolor abdominal en epigastrio que era mantenido y se acompañaba de fatigas referidas. Se solicitó interconsulta con gastroenterología.
Antecedentes patológicos personales de deformidades esqueléticas en miembros inferioresy escoliosis.
Antecedentes patológicos familiares tío paterno y primo hermano con características fenotípicas de síndrome de Marfán.
Se realizan estudios de:
Genética clínica: la evaluación clínica corresponde a un síndrome de Marfán.
Cariotipo: 46xy (14 metafases), masculino cromosómicamente normal.
Endoscopia con biopsia: úlcera duodenal activa con gastritris antral eritemato-nodular, por lo que se decidió realizar el ingreso.
Durante este ingreso comenzó con coloración amarilla ligera de la piel y las escleróticas que desapareció en menos de 48 horas.
Se le realizó:
Glutamato Piruvato Transaminasa: 17,9 u/I.
Glutamato-Oxalacetato Transaminasa: 9,6 u/I.
Gamma Glutamil Transferasa:15,2 u/I.
Bilirrubina T: 90
D: 6,8
I: 83,2
El ultrasonido (UTS) abdominal mostró hígado que sobrepasaba los 2 cm. el reborde costal derecho, impresion óptosishepática, con ecopatrón homogéneo. Vesícula, páncreas, vías biliares y bazo normales. Riñón derecho: medía 102x34 mm, con parénquima de 9,5 mm. Sin litiasis ni hidronefrosis.
Riñón izquierdo: medía 95 x 40 mm, con parénquima de 9 mm, se observó una discreta dilatación calicial del 1/3 superior del uréter (10 mm). Vejiga insuficiente.
Se planteó entonces la posibilidad de una enfermedad de Gilbert.
Se solicitó interconsulta con urología por una pielectasia izquierda, el paciente fue evaluado y se orientó seguimiento trimestral por UTS.
En su evolución posterior ha presentado ictericia ligera relacionada con estrés que desaparece en un corto periodo de tiempo.
Estudios realizados:
Rayos x (Rx) de tórax: No se mostraban lesiones pleuropulmonares. Área cardiaca normal.
La tomografía axial computarizada (TAC) cráneo era normal.
Hormona Folículo Estimulante (FSH): 1,23 muI/lT3 libre 7,40pmol/l.
Hormona Luteilizante (LH): 3,84muI/lT4 libre13,22pmol/l.
Se le realizó ecocardiograma a los 13 años que mostró el corazón estructuralmente sano.
A los 15 años de edad aparece un segundo ruido desdoblado constante en foco aórtico, se evaluó con ecocardiograma nuevamente donde se informó una insuficiencia aórtica ligera, se reevaluó en el Cardiocentro William Soler. Actualmente el paciente continúa con seguimiento semestral, además lleva seguimiento por desnutrición proteico-energética.
Datos positivos al examen físico:
Mucosas coloreadas.
Piel de la espalda: hubo presencia de estrías.
Talla/edad por encima del 97 percentil.
Peso: 55 Kg. Talla: 188 cm.Índice de masa corporal (IMC)/E 15,7, por debajo del 3er percentil.
Paladar ojival.
Sistema osteomioarticular:
Mostró dolicostenomelia (extremidades elongadas en relación a las dimensiones del tronco).
Signo de la muñeca positivo (signo de Walker Murdoch positivo).
Signo del pulgar positivo (signo de Gowers positivo).
Deformidad torácica (pectum excavatum ligero).
Asimetría de los pectorales
Hiperlaxitud articular
Pie plano bilateral.
Examen oftalmológico con trastornos de refracción.
Tratamiento actual:
El paciente toma losartán (50mg) ½ tableta c/ 8h para las alteraciones cardiovasculares. Se mantiene con restricción del ejercicio físico, debe evitar la fatiga. Sigue con vitaminoterapia, dieta hiperproteica, normograsa e hiposódica y con seguimiento multidisciplinario con las especialidades de: cardiología, endocrinología, gastroenterología, fisiatría, ortopedia, nutrición, oftalmología, urología y genética clínica.
La impresión diagnóstica mostró un síndrome de Marfán, enfermedad de Gilbert, pielectasia izquierda e insuficiencia aórtica ligera.
Caso 1

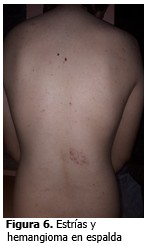
Caso 2

DISCUSIÓN
Las alteraciones cardiovasculares en el Síndrome de Marfán marcan la morbimortalidad de esta afección. Los criterios de Ghent, revisados en 2010 permiten establecer el diagnóstico. En estudio realizado en España a 54 pacientes, según estos criterios, 52 pacientes cumplían criterios de síndrome de Marfán clásico y 2 con síndrome de Marfán neonatal. Se estudió a la mitad de ellos (55,8%, 29/52) con antecedentes familiares conocidos, incluso intraútero. A la otra mitad (44,2%, 23/52), se la diagnosticó por un fenotipo peculiar. De estos, 12 (18,8%) eran casos de novo con estudio genético de ambos progenitores negativo; 10 (15,6%) eran casos índices a partir de los cuales se diagnosticó a algún familiar. De un paciente se desconocían los antecedentes familiares por ser adoptado, presentando el 75% afección familiar.(4)
En la revisión de los dos casos expuestos, ambos clasifican según los criterios de Ghent dentro del síndrome de Marfán clásico, con antecedentes familiares por la línea paterna positivos para esta entidad.
Los pacientes con síndrome de Marfán presentan importantes cambios estructurales cardiacos y eléctricos, así como disfunción ventricular. Dentro de las manifestaciones cardiovasculares en el síndrome de Marfán, por un lado, se encuentran fundamentalmente la insuficiencia aórtica y mitral, los aneurismas y la disección aórtica, esta última causa de un 75-90% de las muertes en esta entidad.(16)
En un estudio de Nicaragua realizado a pacientes con este síndrome las principales alteraciones cardiovasculares diagnosticadas por ecocardiograma fueron las dilatación de la raíz aórtica en el 38% asociado a insuficiencia aórtica en el 10%, el prolapso de la válvula mitral estuvo presente en la cuarta parte de estos niños, la insuficiencia mitral con un porcentaje menor, en un paciente se diagnosticó afección de la aorta ascendente y en otro dilatación de la arteria pulmonar.(17)
El manejo con betabloqueadores incrementa la expectativa de vida conjuntamente con la cirugía vascular y el uso del losartán como alternativa, mientras más temprano comience su uso, menos repercusión cardiovascular, e incluso, se podría evitar el tratamiento quirúrgico.(7)
Ambos pacientes en este trabajo se encuentran con tratamiento con losartán y en el primer caso este fue combinado con cirugía, lo que mejoró su pronóstico.
Conflicto de intereses: los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Contribución de los autores:
Idea conceptual: Elsy Roxana Geroy Moya.
Análisis estadístico: Anaelys Acosta Hernández.
Revisión de la literatura: Elsy Roxana Geroy Moya, María Quiñones Hernández.
Escritura del artículo: Elsy Roxana Geroy Moya, María Quiñones Hernández.
Revisión crítica del artículo: Anaelys Acosta Hernández.
Financiación: Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos.
REFERENCIAS BIBLIOGRÁFICAS
- Cruz KX. Identificación de Nuevas Variantes Genotípicas del Síndrome de Marfán. Un estudio de casos [Internet]. Bogotá: Universidad Distrital Francisco José de Caldas; 2017 [citado 20 Oct 2019]. Disponible en: http://repository.udistrital.edu.co/bitstream/11349/6590/1/CruzMendozaKarenXiomara2017.pdf [Buscar en Google Scholar]
- Carrillo MA, Carreño EH. Síndrome de Marfan. Acércate [revista en Internet]. 2017 [citado 20 May 2018];4(6):[aprox. 10p]. Disponible en: https://portalweb.uacm.edu.mx/uacm/Portals/13/Numeros/6/09 MARFAN.pdf [Buscar en Google Scholar]
- Fuentes MA. Diagnóstico y manejo de las alteraciones del crecimiento en la infancia. Talla baja y talla alta [Internet]. Madrid: Asociación Española de Pediatría; 2016 [citado 20 May 2019]. Disponible en: https://continuum.aeped.es/files/guias/Material_descarga_unidad_3_endocrinologia.pdf [Buscar en Google Scholar]
- Manchola A, Gran F, Teixidó G, López F, Rosés F, Sabaté A. Síndrome de Marfán y síndrome de Loeys-Dietz en la edad pediátrica: experiencia de un equipo multidisciplinar. Rev Esp Cardiol [revista en Internet]. 2018 [citado 12 Feb 2019];71(7):[aprox. 5p]. Disponible en: https://www.revespcardiol.org/es-sindrome-marfan-sindrome-loeys-dietz-edad-articulo-S0300893217302269 [Buscar en Google Scholar]
- Muñoz J, Saldarriaga W, Isaza de Lourido C. Síndrome de Marfán, mutaciones nuevas y modificadoras del gen FBN1. latreia. 2014;27(2):206-215 [Buscar en Google Scholar]
- Enciclopedia Orphanet de la Discapacidad. El síndrome de Marfan [Internet]. España: Orphanet; 2016 [citado 10 Mar 2018]. Disponible en: http://www.orpha.net/data/patho/Han/Int/es/Marfan_Es_es_HAN_ORPHA109.pdf [Buscar en Google Scholar]
- Alvarez FS, Llanos FP. Revisión bibliográfica y presentación de un caso: síndrome de Marfán. Rev Facult Cien Méd Univer Cuenca [revista en Internet]. 2016 [citado 29 Sep 2019];34(1):[aprox. 15p]. Disponible en: http://dspace.ucuenca.edu.ec/jspui/bitstream/123456789/25769/1/Alvarez Palacios Franklin Santiago y LLanos Quilli Fernanda Patricia.pdf [Buscar en Google Scholar]
- Venancio M. Síndrome de Marfan. Nascer e Crescer. Porto Dez [revista en Internet]. 2016;25(1):[aprox. 10p]. Disponible en: http://www.scielo.mec.pt/pdf/nas/v25s1/v25s1a02.pdf [Buscar en Google Scholar]
- Pérez J. Prevención de la muerte súbita cardiaca en Pediatría: el papel esencial del pediatra de Atención Primaria. Rev Pediatr Aten Primaria [revista en Internet]. 2015 [citado 8 Nov 2019];17(65):[aprox. 8p]. Disponible en: http://scielo.isciii.es/pdf/pap/v17n65/19_colaboracion-especial2.pdf [Buscar en Google Scholar]
- Nishimura A. 2017 AHA/ACC focused update of the 2014 AHA/ACC guideline for the management of patients with valvular heart disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J ACC. 2017;70(2):252-89 [Buscar en Google Scholar]
- Riverón LJ, Rojas T, Karusagna D, Ricardo S, Concepción P, Manuel L. Prolapso de la válvula mitral, una revisión bibliográfica. 16 de Abril [revista en Internet]. 2017 [citado 12 Ene 2018];55(261):[aprox. 9p]. Disponible en: https://www.medigraphic.com/pdfs/abril/abr-2016/abr16261c.pdf [Buscar en Google Scholar]
- Ekhomu O, Naheed ZJ. Aortic involvement in pediatric Marfan syndrome: A review. Pediatr Cardiol. 2015;36(5):887-95 [Buscar en Google Scholar]
- Vanegas LM, Boter M, Medina M, Carvajal N. Efectos del ejercicio físico en pacientes con síndrome de Marfán. Duazary [revista en Internet]. 2018 [citado 29 Jun 2019];15(3):[aprox. 11p]. Disponible en: https://doi.org/10.21676/2389783X.2424 [Buscar en Google Scholar]
- Flórez LM, Giraldo MA, Calero MM, Tello NC. Efectos del ejercicio físico en pacientes con síndrome de Marfán. Rev Inter Cien Salud [revista en Internet]. 2018 [citado 1 Ene 2019];15(3):[aprox. 10p]. Disponible en: https://dialnet.unirioja.es/servlet/articulo?codigo=6692324 [Buscar en Google Scholar]
- Santiago AP, Fernanda LQ. Revisión bibliográfica y presentación de un caso: síndrome de Marfán. Rev Facul Cien Méd Cuenca [revista en Internet]. 2016 [citado 10 May 2018];34(1):[aprox. 7p]. Disponible en: https://core.ac.uk/download/pdf/81602874.pdf [Buscar en Google Scholar]
- Pérez P, Carvalho A, Pérez A, Medeiros W. Abnormal heart rate recovery and deficient chronotropic response after submaximal exercise in young Marfan syndrome patients. Cardiol Y. 2016;26(7):1274-81 [Buscar en Google Scholar]
- Velez JU. Comportamiento clínico y epidemiológico del síndrome de Marfán en pacientes pediátricos atendidos en la consulta externa de genética del Hospital Infantil Manuel de Jesús Rivera, Managua. Enero a diciembre 2018. Diss [Internet]. Managua: Universidad Nacional Autónoma de Nicaragua; 2019 [citado 1 Ene 2020]. Disponible en: http://repositorio.unan.edu.ni/11307/ [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129