
Presentaciones de casos
Formas atípicas de presentación de la fibrosis quística: edemas, anemia y lesiones en la piel
Cystic Fibrosis Atypical Forms Presentation: Edema, Anemia and Skin Lesions

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2020-08-13 10:24:54
Aprobado: 2021-04-20 12:02:55
Correspondencia: Chabelys de la Caridad Rodríguez Membrides. Instituto Superior de Ciencias Médicas de Villa Clara. Villa Clara. chamembri97@nauta.cu
RESUMEN
Palabras clave: fibrosis quística; informes de casos
ABSTRACT
Key words: cystic fibrosis; case reports
INTRODUCCIÓN
La fibrosis quística (FQ) o mucoviscidosis es la enfermedad genética más frecuente en la raza caucasiana(1) caracterizada por el espesamiento de las secreciones debido al transporte anormal de iones de las células epiteliales.(2)
La fibrosis quística es además una enfermedad multisistémica, potencialmente fatal. Se caracteriza por una alteración genética en la proteína del gen regulador de la conductancia transmembrana de fibrosis quística. La afectación pulmonar es la más frecuente y la principal causa de mortalidad. La detección neonatal por medio de la tripsina imunoreactiva de posibles casos de fibrosis quistica, ha logrado la detección temprana de los pacientes y para realizar la confirmación diagnóstica con la prueba del sudor y pruebas de ADN. Los moduladores del gen regulador de la conductancia transmembrana de fibrosis quística son los tratamientos más novedosos aprobados para tratar esta enfermedad y tienen como diana el defecto que genera dicha alteración genética, según platea Charpentier Molina en su artículo: Paciente pediátrico con fibrosis quística publicado en la Revista Médica Sinergia.
La incidencia reportada es de 1/3500 recién nacidos vivos en caucásicos y de 1/8000 en hispanos.(3,4)
La FQ se manifiesta de modo habitual con insuficiencia pancreática, enfermedad pulmonar progresiva, alteración en la secreción de electrólitos en sudor, azoospermia en los varones y disminución de la fertilidad en el sexo femenino. Actualmente se acepta que las secreciones espesas son las causantes de la forma clínica que adopta esta entidad nosológica.(4,5)
El tratamiento de la fibrosis quística (FQ) es fundamentalmente sintomático, dirigido a interrumpir el ciclo de retención de moco, infección e inflamación, debe ser de inicio precoz para evitar daños permanentes en el pulmón.(6)
La determinación de electrólitos en el sudor, o prueba de sudor, es la principal herramienta para el adecuado diagnóstico de la FQ desde la publicación en 1959 por Gibson y Cooke del método de iontoforesis con pilocarpina, este método recibió el nombre de Quantitative Pilocarpine Iontophoresic Test (QPIT) (por sus siglas en inglés). Este medio diagnóstico consta de tres etapas: estimular la sudoración, recoger el sudor, y por último, determinar la concentración de electrólitos.(4)
Según Llull Tombo y cols. en su estudio: Caracterización de pacientes con fibrosis quística en consulta multidisciplinaria en el que se realiza una caracterización de los pacientes con fibrosis quística de la provincia Cienfuegos, esta es una enfermedad presente en nuestro medio, con manifestación y complicaciones de inicio temprano. Las características clínicas y epidemiológicas conservan su presentación clásica. La sospecha clínica temprana del diagnóstico de fibrosis quística contribuye a mejorar la calidad de vida del paciente y el tiempo de sobrevida.
Se presenta este caso con el objetivo de describir las características clínicas y de laboratorio de un paciente con presentación atípica de fibrosis quística. Las alteraciones cutáneas como signo de presentación de la fibrosis quística son raras y solamente se han reportado menos de 30 casos en la literatura médica.
PRESENTACIÓN DEL CASO
Se presenta el caso de un lactante de dos meses y medio producto de un embarazo de alto riesgo obstétrico, por embarazo anterior con neonato fallecido el 1er día de nacido por incompatibilidad del sistema ABO. Este paciente nació de parto distócico por cesárea, institucionalizada a las 38,1 semanas con peso al nacer de 3200 gramos, una talla de 52 cm, circunferencia cefálica (cc) 34cm y ct 32cm, con un Apgar 8/9. Presentó íctero fisiológico agravado para lo cual necesitó fototerapia durante 4 días. Tuvo antecedentes de un ingreso previo al mes de vida por presentar una infección del tracto urinario para lo cual llevó tratamiento con claforán. A los 15 días de nacido comenzó a presentar vómitos en número de 2 diarios que se fueron intensificando hasta más o menos 6 de moderada cantidad, con restos de leche, en cualquier momento del día. Un mes después comenzó con lesiones eritematosas irregulares, diseminadas y descamadas en la piel a nivel de la cara, tórax, miembros, región anal y genital, además de presentar poca ganancia de peso. Por estos motivos se decidió su ingreso en el Hospital Pediátrico Universitario José Luis Miranda para estudio y tratamiento.
Estando en sala fue valorado por la especialidad de dermatología donde se describieron lesiones eritematosas generalizadas en región inguinal, abdominal y en las extremidades y la cara.
Examen físico
Al examen físico se observó que el paciente presentaba un estado general conservado, palidez cutáneo-mucosa, lesiones eritemato-papulocostrosas en la piel a nivel de cara, tronco y extremidades. En días posteriores al ingreso se constató la aparición de edemas generalizados, manteniéndose las lesiones en la piel y palidez. (Figs 1 y 2).
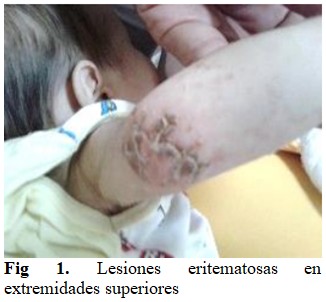
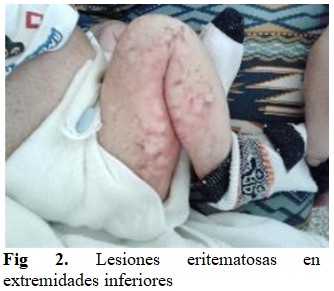
Se auscultó murmullo vesicular audible, sin aleteo nasal, ni tiraje. No se auscultaron estertores. Con frecuencia respiratoria (FR): 35x´.
En el análisis del aparato cardiovascular se reconocieron ruidos cardíacos rítmicos de buen tono e intensidad. Se auscultó soplo sistólico II/VI en región infraclavicular izquierda. Con frecuencia cardiaca (FC): 136x´.
Se observó abdomen suave, depresible, no distendido, sin visceromegalia, ruidos hidroaéreos presentes y conservados. Lactante activo, llanto fuerte, fontanela anterior normotensa.
En la evaluación nutricional al ingreso se constató:
Peso: 4,5 Kg. Talla: 59 cm. Peso/talla: 3-10p. Peso/edad: 25p. Talla/edad: 50-75p.
Exámenes complementarios indicados:
Hemoglobina (Hb): 78g/l. Hematocrito (Hto): 0,25. Leucograma: 8.2x109/l.
Células polimorfonucleares (PMN): 0.40.
Linfocitos: 0.57.
Eritro (Eo): 0.03.
M: 0.00.
Alanina-aminotransferasa (TGP): 45U.l.
Transaminasa oxalacética (TGO): 97U.l.
Hierro (Fe) sérico: 7l.
Conteo de plaquetas: 200x109/l. Proteínas totales: 45 g/L. Albumina: 29 g/L. Creatinina: 37 mmol/l. Triglicéridos: 1,5mmol/l. Urocultivo: negativo. Exudado faríngeo: negativo (flora normal). Ultrasonido abdominal y renal: sin alteraciones. Ecografía cardiaca: persistencia del conducto arterioso (PCA) con ligera repercusión hemodinámica por soplo sistólico en región infraclavicular izquierda. Electrolitos en el sudor 1: 191mmol/l eq NaCl. Electrolitos en el sudor 2: 131 mmol/l eq NaCl. Estudio molecular para fibrosis quística: f508del/R553.
Terapéutica:
Previo al diagnóstico se utilizó crema de manzanilla tópica para las lesiones, metoclopramida (gotas) y transfusión con glóbulos rojos.
Posterior al diagnóstico se administraron multivitaminas en gotas, ácido fólico, sulfato de zinc, domperidona en suspensión, cimetidina (tabletas), enzimas pancreáticas (kreon).
Datos positivos al interrogatorio:
Íctero fisiológico agravado, poca ganancia de peso, vómitos desde hacía dos meses que se fueron incrementando hasta el sexto día.
Datos positivos al examen físico:
Palidez cutáneo-mucosa.
Lesiones eritemato-papulocostrosas que se descaman y son irregulares en piel de la nuca, cara, tronco, extremidades, testículos, pene y región anal, con la presencia de edema generalizado.
Resumen sindrómico:
Síndrome hemolinfopoyético de tipo anémico.
Síndrome hidropígeno.
Síndrome dermatológico infeccioso.
Diagnóstico nosológico:
Fibrosis quística: planteable por tratarse de un lactante de aproximadamente 3 meses con anemia, edema generalizado, lesiones en piel eritemato-papulocostrosas además de poca ganancia de peso. La presencia de vómitos desde los 15 días de vida se interpretó como reflujo gastroesofágico, común en pacientes con esta entidad nosológica. El íctero fisiológico agravado al momento del nacimiento es frecuente en pacientes fibroquísticos.
DISCUSIÓN
Los resultados obtenidos en la investigación de Castilla y cols. con respecto al género se relacionan con los encontrados por Chaustre, en los cuales expresan que la mayoría de sus pacientes con FQ también son de sexo masculino(6) mientras que en la investigación de Barrios y cols. el 50 % de los pacientes con diagnóstico confirmado de fibrosis quística en la Costa Caribe son hombres y el 50 % restante mujeres.(7) Estos resultados coinciden con los presentados en este caso al tratarse de un paciente del sexo masculino.
Sabillón Ortega recoge en su estudio que la edad al momento del diagnóstico en el 60 % de los pacientes fue posterior al 1er año de vida,(8,9,10) aspecto que es diferente a lo encontrado, pues en este estudio fue diagnosticado antes del primer año de edad.
La FQ se manifiesta en su forma clásica y más habitual por enfermedad pulmonar obstructiva crónica, insuficiencia pancreática exocrina (IP), elevación de cloro en sudor e infertilidad en varones por azoospermia obstructiva. Presentaciones menos frecuentes incluyen pacientes con suficiencia pancreática (SP) y algunos casos raros con niveles normales de electrólitos en sudor y con afectación pulmonar leve. Algunos niños presentan solo hipoproteinemia, edema y anemia, lo que se asocia a una alta morbimortalidad.(11,12)
La anemia se ha descrito en relación con las deficiencias nutricionales y hematológicas asociadas con la FQ. El retraso ponderoestatural, la desnutrición y algunas carencias específicas de nutrientes son manifestaciones resultantes de la mala absorción propia de los pacientes fibroquísticos, debido a la IP. La tríada anemia-hipoalbuminemia-edema puede constituir la forma de presentación de la enfermedad en niños pequeños o las alteraciones cutáneas debido a la no absorción de vitamina A, que se asemejan a la acrodermatitis enteropática (causada por deficiencia de Zinc).(13,14)
Las manifestaciones cutáneas de la FQ incluyen erupciones eritematoescamosas, costrosas, psoriasi-formes y/o vesiculoampollares que afectan más acentuadamente a la zona distal de las extremidades y el área del pañal. En el presente caso, el paciente presentó dichas lesiones, sin antecedentes previos de alteraciones respiratorias, presentación habitual de dicha patología. Las alteraciones cutáneas como signo de presentación de FQ son raras y solamente se han reportado menos de 30 casos en la literatura médica.(7,8)
La FQ debe considerarse en el diagnóstico diferencial de niños malnutridos y exantema cutáneo que presentan cuadro sugerente de acrodermatitis enteropática y desnutrición proteico-calórica asociada. La determinación de electrólitos en el sudor, o prueba de sudor, es la principal herramienta para el adecuado diagnóstico de la FQ. Se consideran como valores patológicos compatibles con FQ ≥ 80 mmol/L. El paciente presentó un valor de electrolitos en sudor de 191 mmol/l en el primero y 131 mmol/l en el segundo, quedando corroborado el diagnóstico de dicha entidad.(10)
Conflicto de intereses:
Los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Chabelys Rodríguez Membrides, Carlos Enrique Cruz Carrazana.
2. Curación de datos: Carlos Enrique Cruz Carrazana.
3. Análisis formal: Carlos Enrique Cruz Carrazana.
4. Adquisición de fondos: Esta investigación no contó con la adquisición de fondos.
5. Investigación: Chabelys Rodríguez Membrides, Carlos Enrique Cruz Carrazana, Milda Díaz Martínez.
6. Metodología: Carlos Enrique Cruz Carrazana, Milda Díaz Martínez.
7. Administración del proyecto: Carlos Enrique Cruz Carrazana.
8. Recursos: Chabelys Rodríguez Membrides.
9. Software: Chabelys Rodríguez Membrides.
10. Supervisión: Carlos Enrique Cruz Carrazana.
11. Validación: Milda Díaz Martínez.
12. Visualización: Chabelys Rodríguez Membrides.
13. Redacción del borrador original: Carlos Enrique Cruz Carrazana, Milda Díaz Martínez
14. Redacción revisión y edición: Carlos Enrique Cruz Carrazana, Milda Díaz Martínez.
REFERENCIAS BIBLIOGRÁFICAS
- Ruiz L. Cardiopatía de la fibrosis quística: estudio descriptivo y de posibles factores asociados: Una forma nueva de enfermedad de Keshan [Internet]. Madrid: Universidad Autónoma de Madrid; 2012 [citado 3 May 2018]. Disponible en: https://dialnet.unirioja.es/servlet/tesis?codigo=32645 [Buscar en Google Scholar]
- Pavón D. Manejo nutricional de la fibrosis quística en el paciente pediátrico [Internet]. México: Universidad Autónoma del Estado de México; 2014 [citado 3 May 2018]. Disponible en: https://ri.uaemex.mx/handle/20.500.11799/14720 [Buscar en Google Scholar]
- Fielbaum O. Manejo actual de la fibrosis quística. Rev Méd Clín Las Condes [revista en Internet]. 2017 [citado 6 Ene 2018];28(1):[aprox. 11p]. Disponible en: https://www.sciencedirect.com/science/article/pii/S0716864017300159 [Buscar en Google Scholar]
- Giraldo A, Guerrero R, Saballet B. Caracterización de los pacientes con fibrosis quística de 6 a 18 años en una fundación de Cartagena desde una perspectiva de la CIF [Internet]. Cartagena de Indias: Universidad de Buenaventura; 2012 [citado 4 May 2018]. Disponible en: https://bibliotecadigital.usb.edu.co/bitstream/10819/4458/1/Caracterizacion de los pacientes fibrosis quística_Ana L Giraldo C_2012.pdf [Buscar en Google Scholar]
- Salcedo A, Gartner S, Girón RM, García MD. Tratado de Fibrosis Quística [Internet]. España: Editorial Justim SL; 2012 [citado 8 Nov 2018]. Disponible en: https://www.aeped.es/documentos/tratado-fibrosis-quistica [Buscar en Google Scholar]
- Pizarro ME, Espinoza T. Tratamiento de fibrosis quística: pasado y presente. Neumol Pediatr [revista en Internet]. 2016 [citado 30 Ene 2018];11(1):[aprox. 6p]. Disponible en: https://www.neumologia-pediatrica.cl/wp-content/uploads/2017/07/tratamiento-fq.pdf [Buscar en Google Scholar]
- Barrios WJ, Altamar HS, Jiménez OE, Stand IG, Villamil WE. Caracterización de los pacientes con fibrosis quística en la Costa Caribe colombiana en el periodo comprendido entre 2005 y 2012. Rev Méd Evidencias [revista en Internet]. 2014 [citado 10 Ene 2018];3(1):[aprox. 5p]. Disponible en: https://www.husincelejo.gov.co/pub/UNIDADDEDOCENCIA/REVISTAVOLIIINUM1/8.ARTICULOORIGINAL.pdf [Buscar en Google Scholar]
- Gale S, Sabillón M, Ortega JC. Caracterización de los pacientes con fibrosis quística diagnosticados por cloruros en sudor. Act Pediátr Hondur [revista en Internet]. 2015 [citado 19 Mar 2019];6(2):[aprox. 8p]. Disponible en: https://www.lamjol.info/index.php/PEDIATRICA/article/view/3539 [Buscar en Google Scholar]
- Fuentes G, Portuondo R. Caracterización de pacientes fibroquísticos fallecidos en el curso de su enfermedad. Rev Cubana Pediatr [revista en Internet]. 2014 [citado 8 Nov 2017];86(3):[aprox. 9p]. Disponible en: https://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0034-75312014000300009 [Buscar en Google Scholar]
- Cobos AS, Moscoso MG. Características de la fibrosis quística en pacientes que acuden al Hospital Vicente Corral Moscoso Cuenca-Ecuador 2014-2016 [Internet]. Ecuador: Universidad de Cuenca; 2017 [citado 14 Abr 2018]. Disponible en: https://dspace.ucuenca.edu.ec/handle/123456789/28266 [Buscar en Google Scholar]
- Barón FJ. Descripción de las características clínicas y para-clínicas de pacientes con fibrosis quística en Cartagena de Indias [Internet]. Colombia: Universidad de Cartagena de Indias; 2014 [citado 3 May 2018]. Disponible en: https://repositorio.unicartagena.edu.co/handle/11227/3706 [Buscar en Google Scholar]
- Aquino R, Protzel A, Rivera J, Abarca H, Dueñas M, Nestarez C, et al. Frecuencia de las mutaciones más comunes del gen CFTR en pacientes peruanos con fibrosis quística mediante la técnica ARMS-PCR. Rev Perú Méd Exp Salud Pública [revista en Internet]. 2017 [citado 14 Abr 2018];34(1):[aprox. 7p]. Disponible en: https://www.scielo.org.pe/scielo.php?script=sci_arttext&pid=S1726-46342017000100009 [Buscar en Google Scholar]
- Lentini ER, Navarta LM, López A. Diagnóstico genético de la fibrosis quística: Una experiencia provincial de 10 años, Mendoza, Argentina. Rev Asoc Méd Argen [revista en Internet]. 2017 [citado 14 Abr 2018];130(2):[aprox. 10p]. Disponible en: https://www.ama-med.org.ar/descargacontenido/251 [Buscar en Google Scholar]
- Sojo A, Martínez N, Bousoño C, García MD, Heredia S, Manzanares, et al. Pancreatitis en la fibrosis quística: correlación con el genotipo y estado pancreático. An Pediatr [revista en Internet]. 2011 [citado 3 May 2018];75(6):[aprox. 7p]. Disponible en: https://www.sciencedirect.com/science/article/pii/S1695403311003481 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129