
Artículos originales
Enfermedades esqueléticas de causa genética: experiencia en un servicio de referencia nacional
Skeletal Diseases of Genetic Cause: Experience in a National Reference Service

Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2020-03-10 14:03:45
Aprobado: 2021-01-06 00:07:27
Correspondencia: Estela Morales Peralta. Facultad de Ciencias Médicas 10 de Octubre. La Habana. fornaris@infomed.sld.cu
RESUMEN
Objetivo: analizar aspectos de la clasificación actual de las enfermedades esqueléticas de causa genética, teniendo en cuenta el diagnóstico de los pacientes atendidos en el Servicio de referencia nacional de Genética Clínica.
Métodos: se realizó un estudio descriptivo, retrospectivo, por medio de revisión documental de los datos inscritos entre septiembre de 1984 y diciembre del 2019 en el Servicio de Genética Clínica del Hospital Pediátrico de Centro Habana. Fueron incluidos 225 casos con evidencias clínicas de enfermedades esqueléticas. Se aplicó el método clínico estandarizado en la red de genética. El diagnóstico se realizó utilizando el método comparativo. Las enfermedades identificadas se contrastaron con las incluidas en la clasificación descrita en el 2010.
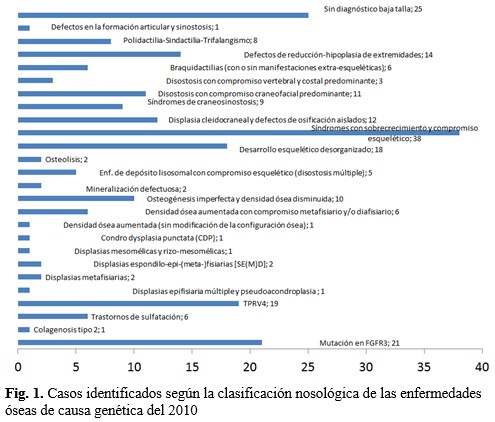
Resultados: se identificó en 190 pacientes enfermedades esqueléticas específicas, las más frecuentes halladas fueron: el síndrome Ehlers-Danlos (n=19; 10 %), la acondroplasia (n=18; 9,4 %) y el síndrome de Marfán (n=17; 8,9 %). De los 40 grupos incluidos en la clasificación analizada, el de mayor número fue el que se correspondía con el sobrecrecimiento. Se halló un paciente con síndrome Wildervanck y un caso con hipoplasia glútea con sinbraquidactilia ipsilateral de miembro inferior, estas enfermedades no están listadas en la clasificación actual.
Conclusión: la clasificación actual de las enfermedades óseas genéticas es producto del conocimiento humano y deberá estar sujeta a cambios.
Palabras clave: enfermedades del desarrollo óseo; enfermedades genéticas congénitas
ABSTRACT
Objective: to analyze aspects of the current classification the skeletal diseases of genetic cause, taking into account the diagnosis of patients treated in a national reference service of Clinical Genetics.
Methods: a descriptive, retrospective study, was carried out by means of a documentary review of the data registered between September 1984 and December 2019 in the Clinical Genetics Service of the Centro Habana Pediatric Hospital. 225 cases with clinical evidence of skeletal diseases were included. The standardized clinical method was applied in the genetics network. The diagnosis was made using the comparative method. The diseases identified were contrasted with those included in the classification described in 2010.
Results: specific skeletal diseases were identified in 190 patients, the most frequent found were Ehlers-Danlos syndrome (n = 19; 10 %), achondroplasia (n = 18; 9.4 %) and Marfan syndrome (n = 17; 8.9 %). Of the 40 groups included in the classification analyzed, the one with the highest number was due to overgrowth. We found a patient with Wildervanck syndrome and a case with gluteal hypoplasia with ipsilateral lower limb synbrachydactyly, these diseases are not listed in the current classification.
Conclusion: the current classification of genetic bone diseases is the product of human knowledge and should be subject to change.
Key words: bone diseases development; genetic diseases inborn
INTRODUCCIÓN
El Servicio de Genética Clínica del Hospital Pediátrico de Centro Habana se estableció en 1984, como parte de la creación de su área de consultas externas. Ha sido desde sus inicios consulta de referencia nacional del Programa Nacional de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos de Cuba, referida en el Programa Asistencial Posnatal para el Diagnóstico Clínico de Enfermedades Genéticas (EECG)(1) del cual forma parte la atención de las enfermedades esqueléticas de causa genética.
Las enfermedades esqueléticas de causa genética son un grupo heterogéneo de afecciones del crecimiento y desarrollo del hueso y el cartílago que incluye más de 350 entidades(2) que individualmente resultan poco frecuentes, pero en su conjunto tienen una incidencia estimada en 1 de 4000 recién nacidos vivos. Por tanto, constituyen un grupo importante de las conocidas como enfermedades raras.(2)
El incremento del conocimiento de las EECG ha conllevado a que su nomenclatura haya sido revisada por expertos en varias ocasiones, en las que se han propuesto clasificaciones basadas en criterios radiológicos, clínicos y según el modo de trasmisión hereditaria. Los radiológicos se basan en la porción del esqueleto afectada, ya sea: epífisis, metáfisis, diáfisis, columna, cráneo, proponiéndose su designación por las partes óseas involucradas. El clínico toma en cuenta principalmente el segmento de las extremidades que esté más acortado, sea el proximal (rizomelia), el intermedio (mesomelia) o el distal (acromelia); según sea la trasmisión hereditaria del patrón de herencia mendeliano que se muestre en la familia.(3,4,5)
Con el advenimiento de nuevas técnicas de genética molecular, la secuenciación del exoma se está convirtiendo en una prueba integrada a la asistencia médica, lo que implica una mayor precisión diagnóstica, y por tanto, la posibilidad de brindar un adecuado asesoramiento genético, con mejores oportunidades terapéuticas.
La clasificación de las enfermedades esqueléticas más actualizada tiene en cuenta las similitudes patogénicas, con base fundamental en los hallazgos moleculares. Sin embargo, el grupo de expertos que la propuso apuntan que esta clasificación, si bien se basa en los criterios moleculares, tiene como propósito principal brindar una lista de referencia y solo secundariamente ayudar al proceso diagnóstico, por lo tanto, requiere coexistir con otras clasificaciones tradicionales basadas en criterios clínicos y radiológicos, como lo es la clasificación de los trastornos constitucionales del hueso, o la clasificación de París y está abierta a contribuciones, especialmente las que se basan en las aplicación de las herramientas clínicas tradicionales.(6)
El objetivo de este trabajo es analizar aspectos de la clasificación actual de las enfermedades esqueléticas de causa genética, teniendo en cuenta el diagnóstico de los pacientes atendidos en el Servicio de referencia nacional de Genética Clínica.
MÉTODOS
Se realizó un estudio descriptivo, retrospectivo, por medio de la revisión documental de los datos inscritos entre septiembre de 1984 y diciembre del 2019 en el Servicio de Genética Clínica del Hospital Pediátrico de Centro Habana. A partir de un total de 6 093 casos registrados, se estudiaron 205 con evidencias clínicas de enfermedades esqueléticas. El diagnóstico se realizó utilizando el método comparativo o de patrón, en este se aplicó el método clínico estandarizado en la red de Genética. Se aplicaron como herramientas:
1. La anamnesis: que se dirigió a obtener los datos generales, la historia de la enfermedad actual, con énfasis en la historia del crecimiento, el árbol genealógico, con al menos, tres generaciones.
2. El examen físico con la búsqueda intencionada de signos clínicos dismórficos y la antropometría.
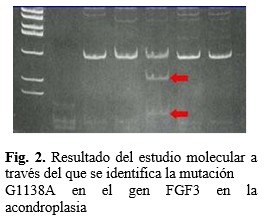
3. Investigaciones individualmente consideradas necesarias. Estas incluyeron fundamentalmente: cariotipo, pruebas metabólicas en orina, estudios imagenológicos, y para los casos en que existían los criterios necesarios, estudio molecular para la búsqueda de la mutación G1138A.
Las enfermedades identificadas se contrastaron con las incluidas en la clasificación descrita en el 2010(3) y se describieron los resultados obtenidos en frecuencias relativas.
El presente estudio está basado en el análisis de los datos de los registros existentes en la consulta de referencia nacional del Hospital Pediátrico de Centro Habana. Los datos de identificación de los pacientes fueron oportunamente eliminados, de acuerdo a las normas éticas de investigaciones en humanos. Fue aprobado por el Comité de Ética e Investigación de la Facultad de Ciencias Médicas de Diez de Octubre de La Habana.
RESULTADOS
Fueron atendidos un total de 6 093 pacientes, en 225 (3,7 %) de ellos se presumió algún tipo de enfermedad esquelética, en 190 se concluyó el diagnóstico específico. Las enfermedades más frecuentes halladas fueron: el síndrome Ehlers-Danlos (n=19; 10 %), la acondroplasia (n=18; 9,4 %), el síndrome de Marfán (n=17; 8,9 %), la neurofibromatosis 1 (n=15; 7,8 %), displasia cleidocraneal (n=12, 6,3 %), el síndrome de Sotos (n=10; 5,2 %), el espectro facioaurículovertebral (n=9, 4,7 %) y la trombocitopenia con ausencia de radios (Síndrome TAR) (n=7; 3,6 %). El resto de las enfermedades correspondieron a menos del 1,5 % de los diagnósticos concluidos.
De los 40 grupos incluidos en la clasificación de enfermedades esqueléticas, el que tuvo mayor número fue el de Síndrome con sobrecrecimiento y compromiso esquelético (Grupo 30), seguido del Grupo 1, debido a cambios del gen FGFR3. (Fig. 1).
En ellos se logró identificar la mutación específica causal en 16 casos, correspondiente a la G1138A. (Fig.2).
Con respecto a su causa, la mayoría fueron monogénicos de transmisión más frecuente autosómica dominante (55 % del total).
Por su importancia es pertinente describir signos clínicos de dos casos que no pudieron ser asignados a ninguno de los grupos de la clasificación vigente:
- Caso 1: se halló anomalía de Klippel-Feil, parálisis del motor ocular externo con retracción del globo ocular y estrechamiento de la fisura palpebral del ojo afectado en la aducción) y sordera neurosensitiva.
- Caso 2: mostró como signos positivos: parálisis facial derecha congénita, limitación de los movimientos laterales de los globos oculares, hipoplasia del glúteo derecho, sinbraquidactilia del pie derecho y pérdida auditiva moderada. En el resto del examen físico no se hallaron otros signos dismórficos relevantes.
El diagnóstico del primero de estos casos fue asignado al grupo 35 (disostosis con compromiso vertebral y costal predominante) y el segundo al 38 (defectos de reducción-hipoplasia de extremidades) de la clasificación actual.
DISCUSIÓN
Los defectos del desarrollo que afectan el esqueleto constituyen un grupo muy heterogéneo dentro de las enfermedades. Su gran variabilidad clínica, bioquímica e histopatológica hacen complejo su diagnóstico y se hallan entre las llamadas enfermedades poco frecuentes (también llamadas raras). Varios estudios realizados sobre la frecuencia de estas afecciones en pacientes ingresados en hospitales pediátricos han demostrado fundamentalmente estar determinadas genéticamente y que las más frecuentes son las autosómicas dominantes, tal cual se halló en esta investigación.(2,7,8)
Debido a su baja frecuencia el diagnóstico es difícil. Por lo que tradicionalmente se utilizan clasificaciones que permiten orientar su identificación, estas se han modificado a través de los años. Inicialmente se basaron en criterios clínicos, fueran estos derivados de las herramientas clínicas tradicionales (especialmente del examen físico) o de las investigaciones imagenológicas tradicionales (fundamentalmente radiografías); así como la región del cuerpo acortada (fueran tronco o extremidades), la porción de los huesos largos más afectada, el comienzo de la enfermedad y su modo de herencia.(3,4,5,6)
Los recientes avances en genética molecular han permitido disminuir los costos (que incluye el tiempo en que demora) de estudios ómicos, de modo que la secuenciación del exoma se está convirtiendo en una prueba integrada a la asistencia médica, lo que implica una mayor precisión diagnóstica, y por tanto, la posibilidad de brindar un adecuado asesoramiento genético, con mejores oportunidades terapéuticas.(9,10)
Esto ha hecho que los hallazgos de estos estudios se han integrado a las clasificaciones y hacen que se surgiera la clasificación descrita en el 2010.(3)
Las observaciones clínicas referidas a casos descritos, como los que se hallan en esta serie de casos, pueden aún enriquecer el conocimiento de las enfermedades esqueléticas y especialmente la sistematización de su conocimiento que deriva en la clasificación.
El grupo 35, con disostosis con compromiso vertebral y costal predominante, no se incluye en el síndrome Wildervanck.(11) Es interesante que en las clasificaciones que antecedieron a la actual, sí se tomaba en cuenta esta enfermedad en el grupo de las disostosis, esto es porque predominantemente tienen alteración de una región específica.(4) En próximas revisiones es lógico que esta enfermedad vuelva a estar incluida.
Existen pocos informes de casos con hipoplasia glútea y sinbraquidactilia ipsilateral.(12,13,14) Si se propone que el síndrome de Poland (hipoplasia o aplasia pectoral con sinbraquidactilia de la mano) se debe a una alteración en la irrigación sanguínea de la arteria subclavia, es lógico que la presencia de hipoplasia glútea y sinbraquidactilia del pie pueda ser interpretada como su equivalente en miembros inferiores, por afección de la ilíaca externa. En otras palabras, que se trate de la ocurrencia de un proceso similar en miembros inferiores al síndrome de Poland, en los superiores, incluso se ha encontrado coincidencia de ambos cuadros en un mismo individuo.(13) En la clasificación más reciente de enfermedades esqueléticas de causa genética se incluye el síndrome de Poland en el grupo 38, correspondiente a los defectos de reducción-hipoplasia de extremidades.(3) El caso dos justifica tal inserción por presentar, amén de las alteraciones de miembros inferiores (similar a los de los miembros superiores), parálisis facial y de los rectos externos.
La clasificación actual de las enfermedades óseas genéticas es producto del conocimiento humano y deberá estar sujeta a cambios.
Conflicto de intereses:
Los autores declaran la no existencia de conflicto de intereses relacionados con el estudio.
Los roles de autoría:
1. Conceptualización: Estela Morales Peralta, Gretell Huertas Pérez.
2. Curación de datos: Estela Morales Peralta.
3. Análisis formal: Estela Morales Peralta.
4. Adquisición de fondos: Esta investigación no recibió ningún financiamiento externo.
5. Investigación: Estela Morales Peralta, Gretell Huertas Pérez.
6. Metodología: Estela Morales Peralta, Gretell Huertas Pérez.
7. Administración del proyecto: Estela Morales Peralta.
8. Recursos: Estela Morales Peralta, Gretell Huertas Pérez.
9. Software: Gretell Huertas Pérez.
10. Supervisión: Estela Morales Peralta.
11. Validación: Estela Morales Peralta, Gretell Huertas Pérez.
12. Visualización: Estela Morales Peralta, Gretell Huertas Pérez.
13. Redacción del borrador original: Estela Morales Peralta.
14. Redacción revisión y edición: Estela Morales Peralta, Gretell Huertas Pérez.
REFERENCIAS BIBLIOGRÁFICAS
- Marcheco B. El Programa Nacional de Diagnóstico, Manejo y Prevención de Enfermedades Genéticas y Defectos Congénitos de Cuba:1981-2009. Rev Cubana Genet Comunit [revista en Internet]. 2009 [citado Feb 8];3(2-3):[aprox. 18p]. Disponible en: https://bvs.sld.cu/revistas/rcgc/v3n2_3/cuba.pdf [Buscar en Google Scholar]
- Hamosh A, Scott AF, Amberger J, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM). Hum Mutat. 2000;15(1):57-61 [Buscar en Google Scholar]
- Warman ML, Cormier V, Hall C, Krakow D, Lachman R, LeMerrer M, et al. Nosology and Classification of Genetic Skeletal Disorders –2010 Revision. Am J Med Genet A. 2011;155A(5):943-68 [Buscar en Google Scholar]
- International nomenclature of constitutional diseases of bones. Ann Radiol (Paris). 1970;13(7):455-64 [Buscar en Google Scholar]
- Beighton P, Giedion A, Gorlin R, Hall J, Horton B, Kozlowski J, et al. International classification of osteochondrodysplasias. International Working Group on Constitutional Diseases of Bone. Eur J Pediatr. 1992;151(6):407-15 [Buscar en Google Scholar]
- Bonafe L, Cormier V, Hall CH, Lachman R, Mortier G, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2015 revision. Am J Med Gen A. 2015;167(12):2869-92 [Buscar en Google Scholar]
- Donaldson J, Aftab S, Bradish C. Achondroplasia and limb lengthening: Results in a UK cohort and review of the literature. J Orthop. 2015;12(1):31-4 [Buscar en Google Scholar]
- Duarte SP, Rocha ME, Bidondo MP, Liascovich R, Barbero P, Groisman B. Bone dysplasias in 1.6 million births in Argentina. Eur J Med Genet. 2019;62(12):103603 [Buscar en Google Scholar]
- Hsu YH, Kiel DP. Clinical review: genome-wide association studies of skeletal phenotypes: what we have learned and where we are headed. J Clin Endocrinol Metab. 2012;97(10):1958-77 [Buscar en Google Scholar]
- Pauli RM. Achondroplasia: a comprehensive clinical review. Orphanet J Rare Dis. 2019;14(1):1 [Buscar en Google Scholar]
- Suri M. Oxford Medical Databases: London Dysmorphology Database Version 3.0. London Neurogenetics Database Version 3.0. Dysmorphology Photo Library on CD-ROM Version 3.0. J Med Genet. 2002;39(10):782-3 [Buscar en Google Scholar]
- Corona JR, Corona A, Totsuka SE, Corona E. Corroboration of the lower extremity counterpart of the Poland sequence. Clin Genet. 1997;51(4):257-9 [Buscar en Google Scholar]
- Parano E, Falsaperla R, Pavone V, Toscano A, Bolan EA, Trifiletti RR. Intrafamiliar phenotypic heterogeneity of the Poland complex: a case report. Neuropediatrics. 1995;26(4):217-9 [Buscar en Google Scholar]
- Riccardi VM. Unilateral gluteal hypoplasia and brachysyndactyly: lower extremity counterpart of the Poland anomaly. Pediatrics. 1978;61(4):653-4 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129