
Artículos originales
Diagnóstico de aciduria metilmalónica en el periodo 2013-2018
Diagnosis of Methyl Malonicaciduria from 2013 to 2018
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2020-01-30 14:27:09
Aprobado: 2020-02-06 10:19:35
Correspondencia: Alina Concepción Álvarez. Centro Nacional de Genética Médica. La Habana. alinaconcepcion@infomed.sld.cu
RESUMEN
Objetivo: describir la implementación de una metodología de laboratorio que combina el ácido metilmalónico y la homocisteína en el diagnóstico diferencial y seguimiento de la aciduria metilmalónica en el periodo de 2013 a 2018.
Métodos: a los pacientes con incremento de ácido metilmalónico en el perfil de ácidos orgánicos, se les cuantificó homocisteína en plasma y orina. La identificación del ácido metilmalónico se realizó por cromatografía gaseosa/ espectrometría de masas, mientras que la cuantificación de homocisteína por cromatografía líquida de alta resolución.
Resultados: los métodos cromatográficos permitieron la identificación y cuantificación del ácido metilmalónico y la homocisteína, respectivamente. La homocisteína se cuantificó en siete pacientes con niveles incrementados de aciduria metilmalónica. Los niveles de homocisteína en cuatro de ellos fueron superiores a los valores normales, sugiriendo una aciduria combinada con homocistinuria. Tres de los pacientes con aciduria metilmalónica combinada bajo tratamiento mostraron una disminución en los niveles de ambos metabolitos, correspondiendo con una satisfactoria evolución.
Conclusiones: la metodología implementada con los análisis de la determinación simultánea de ambos marcadores permitió el diagnóstico diferencial y seguimiento bioquímico de la aciduria metilmalónica.
Palabras clave: diagnóstico; ácido metilmalónico; enfermedades genéticas congénitas
ABSTRACT
Objective: to describe the implementation of a laboratory methodology that combines methylmalonic acid and homocysteine in the differential diagnosis and monitoring of methylmalonic aciduria in the period from 2013 to 2018.
Methods: for patients with an increase in methylmalonic acid in the organic acid profile, homocysteine was quantified in plasma and urine. The identification of methylmalonic acid was performed by gas chromatography / mass spectrometry, while the homocysteine quantification by high performance liquid chromatography.
Results: chromatographic methods allowed the identification and quantification of methylmalonic acid and homocysteine, respectively. Homocysteine was quantified in seven patients with increased levels of methylmalonic aciduria. Homocysteine levels in four of them were higher than normal values, suggesting aciduria combined with homocystinuria. Three of the patients with combined methylmalonic aciduria under treatment showed a decrease in the levels of both metabolites, corresponding to a satisfactory evolution.
Conclusions: simultaneous determination of both markers allowed differential diagnosis and biochemical monitoring of this disease.
Key words: diagnosis; methylmalonic acid; genetic diseases inborn
INTRODUCCIÓN
La aciduria metilmalónica (AM) es una de las acidurias orgánicas más frecuentes, caracterizadas por aumento de la excreción de ácido metilmalónico (AMM) en orina. Este grupo de enfermedades puede estar dado por mutaciones de la metilmalonil CoA mutasa o alteraciones en la síntesis de la cobalamina. Las AMs se clasifican en aisladas o combinadas, en estas últimas se observa un incremento de los niveles de homocisteína (Hcy) en el plasma.(1,2)
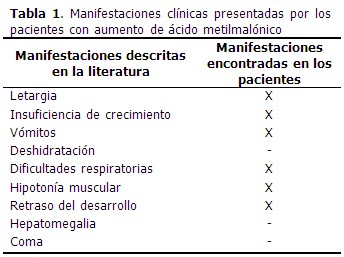
Dentro de los síntomas más frecuentes observados se encuentran la letargia, la insuficiencia de crecimiento, vómitos recurrentes, deshidratación, dificultades respiratorias e hipotonía muscular. Otros signos son el retraso del desarrollo, hepatomegalia y el coma.(3) El tratamiento de los pacientes con AM se basa en la administración de L-carnitina y vitamina B12. Los pacientes pueden ser respondedores o no respondedores al tratamiento con vitaminas. Por lo que requieren de controles bioquímicos y clínicos durante la primera fase de tratamiento.(4,5)
El diagnóstico diferencial de los casos con AM requiere de la aplicación de algoritmos complejos, donde la evaluación de los niveles de AMM y Hcy constituye el primer paso. El análisis cualitativo del perfil de ácidos orgánicos es suficiente para detectar la AM. La evaluación simultánea de los niveles de Hcy permite detectar las variantes combinadas o aisladas de la AM, obteniendo así un diagnóstico más específico.
En Cuba, se realiza el análisis cualitativo de ácidos orgánicos en la pesquisa selectiva de poblaciones de riesgo. Actualmente la AM constituye una de las acidurias orgánicas que con más frecuencia se ha observado.(6) La introducción de la cuantificación de la Hcy nos ha permitido realizar un diagnóstico lo más específico posible y dar seguimiento a los casos identificados con elevaciones del AMM.
El objetivo de este trabajo es describir la implementación de una metodología de laboratorio que combina el AMM y la Hcy en el diagnóstico diferencial y seguimiento de la AM en el periodo de 2013 a 2018.
MÉTODOS
El perfil de ácidos orgánicos se obtuvo a solicitud de los especialistas de todo el país, en pacientes con hallazgos clínicos sugestivos de una aciduria orgánica. En aquellos casos donde se observó un aumento de AMM, se cuantificó la homocisteína en orina o en plasma.
Se utilizaron muestras de orina de la primera micción y muestras de plasma con anticoagulante etilendia- minotetra-acético (EDTA), ambas recolectadas con ayuno superior a 6 horas.
El AMM se detectó en orina, por un método de cromatografía gaseosa acoplada a espectrometría de masas descrito por Camayd y cols.(6) La verificación de la autenticidad de los espectros de masas obtenidos se realizó mediante la comparación de los espectros procedentes de los registros de la base de datos del Instituto Nacional de Estándares de los Estados Unidos (NIST versión 2011) y la base de datos MassBank.(7) Los niveles de AMM se estimaron semi-cuantitativamente usando controles de AMM en agua a 10, 25, 50 y 100 μM. Se consideraron positivas las muestras, donde el área del AMM en el cromatograma es superior a la del control de 10 μM.
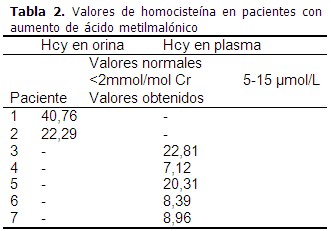
La cuantificación de Hcy en plasma y orina, por cromatografía líquida de alta resolución se realizó a las muestras de los pacientes diagnosticados con AM, utilizando el método descrito por Concepción y col.(8) La cuantificación de la Hcy se realizó sustituyendo el área de este aminoácido en la ecuación de la curva de calibración. Se consideró como valores de referencia los descritos en la base de datos Human Metabolome Database (plasma: 5-15 μM y orina: <2 mmol/mol de creatinina).(9) La creatinina se cuantificó en muestras de orina, utilizando los juegos de reactivos comerciales (HELFA) en el analizador automático Elimat.
RESULTADOS
En el periodo de noviembre de 2013 a diciembre de 2018 se analizaron 450 muestras de pacientes de todo el país. De ellos, 7 presentaron niveles incrementados de AMM: 5 niveles moderados (entre 10 y 100 mmol/mol de creatinina) y 2 niveles superiores a 100 mmol/mol de creatinina.
Las principales manifestaciones clínicas presentadas por los pacientes con aumento de AMM se describen a continuación. (Tabla 1).
La Hcy se cuantificó en los 7 pacientes con aumento de AMM. De estos pacientes 4 presentaron niveles alterados de Hcy. (Tabla 2).
DISCUSIÓN
En este estudio no fue posible cuantificar la Hcy siempre en el mismo tipo de fluido, debido a la disponibilidad de las muestras en cada caso, lo cual constituye una limitación, pues en general se prefiere el plasma para cuantificar este metabolito.(9) No obstante, en los pacientes 1 y 2 pudo demostrarse que tanto la Hcy como el AMM estaban elevados en orina, lo que se traduce en un aumento a nivel plasmático.
El aumento simultáneo del AMM y la Hcy en los pacientes 1, 2, 3 y 5, sugiere una AM combinada con homocistinuria, donde se podría inferir que el defecto se encuentra en el metabolismo intracelular de la cobalamina, específicamente en las variantes cblC, cblD, cblF o cblJ.10 Además, es posible descartar una encefalopatía mitocondrial con AMM elevado (asociado a los genes SUCLA2 y SUCLG1), ya que esta patología cursa con aumento de los niveles de ácido láctico en sangre y orina.(10,11) Los perfiles de ácidos orgánicos no mostraron una aciduria láctica; además, en ninguno de los casos se refieren cuantificaciones alteradas de lactato en sangre. Por otro lado, las variantes aisladas se caracterizan por episodios severos de acidosis, hiperamonemia o cetosis, no referidos en el resumen de historia clínica. Asimismo, el 3-hidroxibutirato y el acetoacetato (cuerpos cetónicos) son detectables en el perfil de ácidos orgánicos, pero no se identificaron en las muestras analizadas.
Una vez diagnosticados los pacientes con AM, solo se pudo evaluar en tres la respuesta al tratamiento con vitamina B12 y L-carnitina. El paciente 1 falleció antes del diagnóstico. En este caso solo pudo definirse una variante de AMM combinada con homocistinuria, sin definir la respuesta a tratamiento con vitamina B12. Los niveles de ambos marcadores sugieren alguna de las variantes cblC, cblD1, cblF o un defecto de malabsorción de vitamina B12.
El seguimiento de los pacientes 2, 3 y 5, después de iniciado el tratamiento con vitamina B12 y L-carnitina intramuscular evidenció una caída drástica en los niveles de AMM y una disminución de los niveles de Hcy (Paciente 2: 4,25 mmol/mol de creatinina, Paciente 3: 3,29 μmol/L, Paciente 5: 8,03 μmol/L), que se correspondió con una evolución satisfactoria. Este hecho sugiere una de las variantes de AM que responden a tratamiento con vitamina B12 (cblC, cblD1, cblF o un defecto de malabsorción de vitamina B12).
En los pacientes 4, 6 y 7 se sugirió una AM aislada, donde el defecto genético podría ser en el gen que codifica para la enzima metilmalonil-CoA mutasa, o en los cofactores de adenosilcobalamina, descritos como variantes aisladas de AM (cblA, cblB y cblD-2).(3,12) En este caso, el perfil de ácidos orgánicos durante la crisis, con aumento del AMM por debajo de 10000 mmol/mol de creatinina y sin aciduria láctica, es sugestivo de variantes donde están deficientes los cofactores de adenosilcobalamina.(12)
El análisis de ambos marcadores bioquímicos permitió realizar el diagnóstico diferencial de la aciduria metilmalónica, así como brindar seguimiento a los pacientes con niveles aumentados. La metodología implementada en nuestro laboratorio, con el análisis del AMM y la Hcy en el diagnóstico diferencial de la AM permite: (1) definir la presencia de una AM aislada o combinada con Hcy, (2) seguir a los pacientes identificados y establecer si la AM responde o no al tratamiento con vitamina B12 y (3) proponer a partir de los datos clínicos y los resultados de laboratorio, las posibles variantes de AM.
Conflicto de intereses: los autores declaran la no existencia de conflictos de intereses relacionados con el estudio.
Contribución de los autores:
Idea conceptual: Alina Concepción Álvarez.
Análisis estadístico: Ivette Camayd Viera, Laritza Martínez Rey.
Revisión de la literatura: Alina Concepción Álvarez, Norma Elena de León Ojeda, Alina García.
Escritura del artículo: Alina Concepción Álvarez.
Revisión crítica del artículo: Daniel Quintana Hernández.
Financiación: Centro Nacional de Genética Médica. La Habana.
REFERENCIAS BIBLIOGRÁFICAS
- Valle D, Antonarakis S, Ballabio A, Beaudet A, Mitchell GA. The Online Metabolic and Molecular Bases of Inherited Disease. New York: McGraw-Hill Education; 2015 [Buscar en Google Scholar]
- Concepción A, Camayd I, León NE, García A, Tamayo V, Quintana D. Introducción de la homocisteína como marcador en el diagnóstico diferencial de la aciduria metilmalónica [Internet]. La Habana: Congreso de Genética Comunitaria; 2017 [citado 26 Sep 2018]. Disponible en: http://geneticacomunitaria2017.sld.cu/index.php/gencom/2017/paper/view/497 [Buscar en Google Scholar]
- ORPHA. Acidemia metilmalónica resistente a la vitamina B12 tipo mut- [Internet]. Mississippi: ORPHA; 2016 [citado 8 Jun 2018]. Disponible en: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=79312&lng=ES [Buscar en Google Scholar]
- Baumgartner MR, Hörster F, Dionisi C, Haliloglu G, Karall D, Chapman KA, et al. Proposed guidelines for the diagnosis and management of methylmalonic and propionic acidemia. Orphanet J Rare Dis. 2014;9(1):130 [Buscar en Google Scholar]
- Horster F, Baumgartner M, Viardot C, Suormala T, Burgard P, Fowler B, et al. Long-term outcome in methylmalonic acidurias is influenced by the underlying defect (mut0, mut-, cblA, cblB). Pediatr Res. 2007;62(2):225-30 [Buscar en Google Scholar]
- Camayd I, Nuevas L, Concepción A. Diagnóstico bioquímico de acidurias orgánicas en Cuba: periodo 2008-2013. Acta Bioquím Clín Latinoam. 2015;49(2):209-14 [Buscar en Google Scholar]
- European MassBank. High Quality Mass Spectral Database [Internet].European MassBank: MassBank Project; 2017 [citado 29 Jun 2018]. Disponible en: https://massbank.eu/MassBank/ [Buscar en Google Scholar]
- Concepción A, Camayd I, Nuevas L. Validación de un método para la cuantificación de homocisteína total en plasma por HPLC. Rev Lab Clin. 2016;9(2):40-7 [Buscar en Google Scholar]
- Human Metabolome Database. The Human Metabolome Database for 2018 [Internet]. Canadá: Canadian Institutes of Health Research; 2018 [citado 27 Nov 2019]. Disponible en: http://www.hmdb.ca/ [Buscar en Google Scholar]
- Sloan JL, Carrillo N, Adams D, Venditti CP. Disorders of Intracellular Cobalamin Metabolism [Internet]. Seattle: University of Seattle; 2008 [citado 23 Oct 2019]. Disponible en: https://www.ncbi.nlm.nih.gov/pubmed/20301503 [Buscar en Google Scholar]
- Haas RRH, Parikh S, Falk MJ, Sanetod RP, Wolfe NI, Darinf N, et al. Enfermedad mitocondrial: abordaje práctico para los médicos. Pediatría [revista en Internet]. 2007 [citado 19 Dic 2019];64(6):[aprox. 10p]. Disponible en: https://www.elsevier.es/es-revista-pediatrics-10-articulo-enfermedad-mitocondrial-abordaje-practico-medicos-13114053 [Buscar en Google Scholar]
- Fowler B, Leonard JV, Baumgartner MR. Causes of and diagnostic approach to methylmalonic acidurias. J Inherit Metab Dis. 2008;31(3):350-60 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129