
Presentaciones de casos
Enfermedad de Castleman en pediatría. Presentación de un caso y revisión de la literatura
Castleman Disease in Pediatrics. Case Presentation and Bibliography Review
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2019-01-12 00:25:56
Aprobado: 2019-01-12 00:32:32
Correspondencia: Lucía Díaz Morejón. Hospital Pediátrico Universitario Paquito González Cueto. Cienfuegos. ldm@hosped.cfg.sld.cu
RESUMEN
Palabras clave: enfermedad de castleman; adolescente; informes de casos
ABSTRACT
Key words: castleman disease; adolescent; case reports
INTRODUCCIÓN
La enfermedad de Castleman (EC), también llamada hiperplasia linfoide angiofolicular, hematoma e hiperplasia gigante de los ganglios linfáticos, angiomatosis linfoidea y tumor gigante benigno linfomatoso, es una rara enfermedad descrita por primera vez en el año 1956, con igual frecuencia en ambos sexos, que puede aparecer a cualquier edad. Se caracteriza por crecimiento de tumores benignos del tejido linfático, que se localizan con mayor frecuencia en mediastino, estómago y cuello, y es menos frecuente en la axila, pelvis y páncreas.(1-5)
Se distinguen dos formas de la enfermedad:
La forma localizada: masa asintomática, aislada, localizada en mediastino o menos frecuente en abdomen, que pueden acompañarse de dolor en relación a la comprensión local del tumor. El tratamiento es la extirpación quirúrgica de la masa.
La forma generalizada o multicéntrica: afecta a más de un área y cursa con síntomas generales, hepato-esplenomegalia, es más frecuente en varones inmunosuprimidos, lo que se asocia a alteraciones analíticas tales como: eritrosedimentación acelerada, trombocitopenia, leucopenia, proteinuria.
Aproximadamente la tercera parte de los casos desarrolla enfermedades malignas como linfoma no Hodking, sarcoma de Kaposi y otros carcinomas.(1-5)
Esta forma es excepcional en la edad pediátrica. Histológicamente se describen 3 variedades: La hialinovascular (80-90 % casos), la plasmocelular (10 %) se relaciona con la forma multicéntrica o diseminada de la enfermedad y la mixta (2 %).
El diagnóstico de este proceso requiere la resección completa del ganglio afectado, no se considera útil la biopsia por aspiración.(6) En las formas localizadas la resección completa del ganglio es curativa. En la forma multicéntrica parecen tener una respuesta más favorable al tratamiento en el niño que en el adulto.
La utilización de radioterapia, esteroides y quimioterapia específica han mejorado el pronóstico de esta variante que todavía presenta un 60 % de mortalidad en la población adulta.(7,8)
Se presenta este reporte por la baja prevalencia de este tipo de tumor en el Hospital Pediátrico Universitario Paquito González Cueto de Cienfuegos.
PRESENTACIÓN DEL CASO
Se presenta el caso de un paciente de 13 años de edad, de color de piel blanca y de procedencia rural. Producto de un embarazo y parto normales. Este paciente tuvo antecedentes de un primer ingreso a los 5 años por síndrome febril prolongado, donde se constató palidez ictérica de la piel y mucosas, bazo e hígado palpables, adenopatías generalizadas, pequeñas, no adheridas a planos profundos sin signos inflamatorios, se diagnosticó una microesferocitosis congénita e infección por citomegalovirus (CMV), con hallazgos del virus en saliva y orina, así como inmunodeficiencia celular por pruebas de inmunidad retardada. Se le orientó tratamiento con factor de transferencia, ácido fólico y multivitaminas, se mantuvo seguimiento en consulta de hematología y a los 9 años se decidió realizar esplenectomía parcial, por aumento de las demandas transfusionales con lo cual se logró estabilizar la hemoglobina mayor de 120 g/l.
A los 11 años ingresó nuevamente con diagnóstico de síndrome febril prolongado para estudio, mantenía buen estado general, pero presentaba palidez cutáneo-mucosa, al examen físico se constató hepatomegalia de 2-3 cm como único dato positivo de interés clínico.
Se realizan complementarios: Hemograma con cifras de hemoglobina normales, conteo de reticulocitos: 50 x10-3 l, Leucocitosis neutrofílica con granulaciones tóxicas en los segmentados, radiografía de tórax, senos paranasales y survey óseo sin alteraciones, transaminasas (TGP, TGO) normales, factor reumatoide y proteína C reactiva positivos, células LE negativas, electroforesis de proteína: elevada la gammaglobulina y disminuida la betaglobulina. Se realizó además test de rosetas disminuido (49 %). Estudios serológicos para hepatitis B y C, Einstein Bar y citomegalovirus negativos y hemocultivos, coprocultivos, urocultivos y bilicultivo negativos.
Se indicó ecografía abdominal que mostró presencia de hepatomegalia de aproximadamente 3 cm, sin cambios ecogénicos a señalar, por lo que se decidió realizar laparoscopia exploradora en la que se constató microabcesos hepáticos, de los cuales se aisló estafilococo aureus y se diagnosticó histológicamente larva migrans visceral y hepatitis reactiva, se prescribió tratamiento específico con antibióticos y antiparasitarios (tiabendazol una tableta de 500 mg), dosis 50 mg/Kg/día). El paciente evolucionó satisfactoriamente para reingresar luego de 3 meses con fiebre de 15 días de evolución, sin otra sintomatología acompañante. Se realizaron nuevamente complementarios: exudados de los cuales mostraron la presencia de bacilos acidorresistentes (BAAR) en un cultivo de esputo y el diagnóstico serológico del virus de la inmunodeficiencia humana (VIH), todos realizados en primera y segunda ocasión fueron negativos. Medulograma: integridad de los tres sistemas, sin la presencia de células ajenas al parénquima medular.
En la ecografía abdominal se observaron adenomegalias peri-aórticas y de hilio hepático, la mayor de los cuales medía de 40x19 cm en hilio-esplénico otra de 47x34 cm; litiasis hiliar única. El tejido esplénico impresionaba formaciones nodulares de tamaño variable, resto normal. (Figura 1).
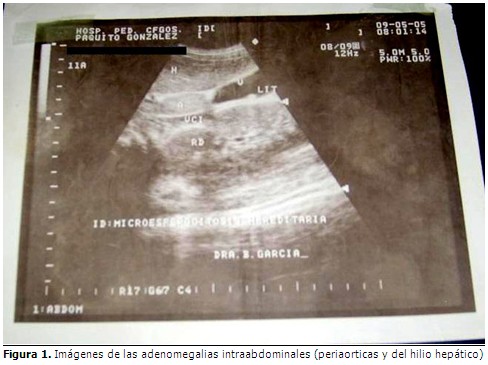
Se realizó tomografía axial (TAC) abdominal simple y contrastada que informó: hígado aumentado de tamaño, bazo pequeño de densidad normal, vesícula, páncreas y riñones normales. Adenopatías periaórticas y de hilio hepático que no se describían.
Se discutió en colectivo y se decidió de conjunto con cirugía realizar laparotomía exploradora donde se constató tejido esplénico con nódulos hiliares múltiples, se realizó exceresis total del bazo, se tomaron muestra de múltiples ganglios intrabdominales y de tejido hepático, vesícula con litiasis única.
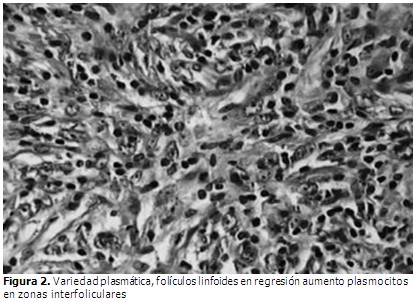
En la biopsia de ganglio linfático intrabdominal se apreció: ganglio linfático con borramiento parcial de su arquitectura con hiperplasia angiofolicular predominante y otros con signos de atrofia, se diagnosticó enfermedad de Castleman con afectación ganglionar difusa además de hepatitis crónica reactiva. (Figura 2).
Actualmente el paciente mantiene buen estado general, se encuentra asintomático, por lo que se mantiene seguimiento ambulatorio con control hematológico y ultrasonido abdominal así como tratamiento con vitaminoterapia.
DISCUSIÓN
La enfermedad de Castleman es un padecimiento raro, del que se ignora su incidencia real, ocurre por igual en hombres y mujeres y no se ha descrito preferencia por alguna raza. Se caracteriza por una hiperplasia no clonal en los ganglios linfáticos.(1-5)
Los tumores benignos del tejido linfático se localizan con mayor frecuencia en el mediastino (interior de la cavidad torácica), estómago y cuello. Los sitios menos comunes incluyen axila, pelvis y páncreas. Se desconoce la causa de la enfermedad, aunque parece existir alguna relación, aún no bien aclarada, con el herpes virus tipo 8 HHV8.
Existen tres tipos histológicos de la enfermedad de Castleman:
- El tipo vascular hialino es el que se presenta más frecuentemente, en alrededor del 90 % de los casos. La mayoría son asintomáticos y localizados o pueden desarrollar crecimientos no cancerosos en los nódulos linfáticos.
- El tipo de células plasmáticas de la enfermedad de Castleman suele asociarse a una forma generalizada y se puede manifestar por fiebre, pérdida de peso, erupciones de la piel, anemia hemolítica, e hipergammaglobulinemia.
- El tipo mixto de la enfermedad de Castleman se caracteriza por compartir de las dos formas anteriormente detalladas.
El paciente del reporte referido presentaba antecedentes de anemia e hiperganmaglobulinemia, manifestaciones sistémicas, lo cual está relacionado con esta variedad histológica de la enfermedad.(6-8)
No se ha descrito un mecanismo definitivo en la patogénesis y progresión de la enfermedad de Castleman. La fisiopatogenia de este padecimiento está marcada por un incremento de la interleucina 6, que a su vez determina un aumento de los niveles del factor de crecimiento endotelial vascular (VEGF), factor de crecimiento epidermal (EGF) e interferón alfa. La presencia del virus herpes 8 y VIH se asocia a enfermedad multicéntrica de Castleman, aunque en la unicéntrica no siempre se detecta al virus herpes 8 en los pacientes y en general son VIH negativos, por lo que se postula que la enfermedad de Castleman unicéntrica y multicéntrica son dos enfermedades diferentes con un nexo fisiopatológico común. No existen detalles patognomónicos que nos permitan sospechar la aparición de la enfermedad. El diagnóstico definitivo es anatomopatológico, la cirugía es curativa, la supervivencia de los pacientes a 5 años es de casi el 100 %, y las recidivas son muy raras.(6-8)
Se presenta un caso con diagnóstico de enfermedad de Castleman con afectación ganglionar difusa, el cual cursó con síndrome febril prolongado recurrente, con diagnóstico histoanatomopatológico del tejido ganglionar con una evolución favorable.
Esta enfermedad puede presentarse como afectación ganglionar difusa, acompañanda de un síndrome febril prolongado recurrente. El diagnóstico definitivo y la curación se realizan solo mediante la exéresis quirúrgica.(6-8)
Se hace indispensable el estudio histológico durante la evaluación de neoplasias ganglionares, así como la sospecha clínica y patológica de las diferentes variedades de enfermedad de Castleman, para permitir un tratamiento oportuno y evitar el deterioro clínico de los pacientes y que estos se diagnostiquen con enfermedad avanzada.(6-8)
REFERENCIAS BIBLIOGRÁFICAS
- López A, Gázquez I, Martín A, Rubio M. Enfermedad de Castleman localizada en región cervical. An Med Interna (Madrid) [revista en Internet]. 2003 [citado 18 Feb 2018];20(12):[aprox. 2p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-71992003001200012&lng=es [Buscar en Google Scholar]
- Coca I, Ortega MV, Fernández E, Gavilán JC, Bermúdez F. Enfermedad de Castleman localizada: descripción de un caso y revisión de la literatura. An Med Interna (Madrid) [revista en Internet]. 2003 [citado 18 Feb 2018];20(10):[aprox. 3p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-71992003001000009&lng=es [Buscar en Google Scholar]
- Rodríguez H, Buchaca E, Machado I, Pérez G, Pérez D. Enfermedad de Castleman: Presentación de cinco casos. An Med Interna (Madrid) [revista en Internet]. 2005 [citado 19 Feb 2018];22(1):[aprox. 4p]. Disponible en: http://scielo.isciii.es/scielo.php?script=sci_arttext&pid=S0212-71992005000100006&lng=es [Buscar en Google Scholar]
- Sánchez de Toledo S, Fábrega C, Marhueenda X, Lucaya N, Torán N, Gross C, et al. Enfermedad de Castleman. Anales de Pediatría [revista en Internet]. 2005 [citado 23 Ene 2017];63(1):[aprox. 4p]. Disponible en: https://www.sciencedirect.com/science/article/pii/S1695403305701429 [Buscar en Google Scholar]
- De Marchi G, De Vita S, Fabris M, Scott CA, Ferracioli G. Systemic connective tissue disease complicated by castleman´s disease: report of a case and review of the literature. Haematologica. 2004;89(4):ECR03 [Buscar en Google Scholar]
- Puerto IM, Martínez IB, Ochoa MC. Folicular dendritic cell sarcoma arising from nodal hyaline-vascular. Castleman´s disease: Case Report. HAEMA. 2003;6(1):81-4 [Buscar en Google Scholar]
- Altiparmak MR, Pamuk ON, Pamuk GE, Dogusny G. Secondary amyloidosis in castleman´s disease: review of the literature and report of a case. Ann Hematol. 2002;81(6):336-9 [Buscar en Google Scholar]
- Avellaneda A, Pérez A, Allez P, Gutiérrez E, Izquierdo M. Percepción de las enfermedades raras por el médico de atención primaria. SEMERGEN [revista en Internet]. 2012 [citado 23 Dic 2018];1(7):[aprox. 11p]. Disponible en: http://www.enfermedades-raras.org/es/default.htm [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129