
Artículos de revisión
Sarcomas: etiología y síntomas
Sarcomas: etiology and symptoms
Especialista de I Grado en Medicina Interna. Profesor Instructor.
Cómo citar este artículo:
Copyright: Esta revista provee acceso libre inmediato a su contenido bajo el principio de que hacer disponible gratuitamente investigación al publico apoya a un mayor intercambio de conocimiento global. Esto significa que se permite la copia y distribución de sus contenidos científicos por cualquier medio siempre que mantenga el reconocimiento de sus autores, no haga uso comercial de las obras y no se realicen modificaciones de ellas.

Recibido: 2012-03-27 18:17:49
Aprobado: 2012-05-04 09:30:51
Correspondencia: Roberto Gabriel Albín Cano. Hospital Clínico-Quirúrgico Freyre de Andrade, Ciudad de La Habana roberto.albin@infomed.sld.cu
RESUMEN
Palabras clave: sarcoma; signos y síntomas; factores de riesgo
ABSTRACT
Due to the wide diversity of sarcomas, almost no texts include all varieties of this type of cancer. Generally, their description and review is included in those of the specifically affected organ system, and the literature containing that information is very fragmented in different medical specialties. We performed a literature review on the etiology and symptoms of most types of sarcomas. It is aimed at achieving a recompilation of most current information available on the causes and symptoms of sarcomas. Different risks and etiologic factors have been identified regarding genetics, infections, and environment. The great discoveries regarding genetic mechanisms involved in different types of sarcomas, have opened an invaluable way to introduce new treatments, including monoclonal antibodies and new drugs of gene therapy.
Key words: sarcoma; signs and symptoms; risk factors
INTRODUCCIÓN
El sarcoma es un cáncer del tejido mesenquimatoso (tejido de sostén), que se caracteriza por tener escaso estroma de tejido conectivo, razón por la que son carnosos (sarco: carnoso).
Tanto los niños como los adultos pueden desarrollar un sarcoma, que puede originarse en cualquier parte del cuerpo, como el tejido óseo o el blando.
Aproximadamente el 60 % de los sarcomas comienza en los brazos o las piernas, el 30 % en el tronco o el abdomen, y el 10 % en la cabeza o el cuello. El sarcoma es poco frecuente y representa alrededor del 1% de todos los casos de cáncer en adultos. Sin embargo, el sarcoma en general, representa aproximadamente el 15 % de todos los casos de cáncer en los niños.
Los sarcomas de tejido blando (STS, por su sigla en inglés: Soft Tissue Sarcoma) son un grupo de tumores malignos que se originan en los tejidos que dan soporte y conexión al cuerpo. Los STS pueden aparecer casi en cualquier parte del cuerpo. Las células del sarcoma se parecen a aquellas que mantienen unido el cuerpo, incluidas las células grasas, los músculos, los nervios, los tendones, las articulaciones, los vasos sanguíneos o los linfáticos. Cuando un STS es pequeño, puede pasar desapercibido, ya que generalmente no causa síntomas al inicio. A medida que crece, puede complicar las funciones corporales normales.1,2
En los Estados Unidos de Norteamérica, la incidencia anual es de aproximadamente 10 400 casos con cerca de 3680 muertes por año.3 El tiempo de supervivencia ante la presencia de estos tumores varía en dependencia del tipo, localización, estadio en que se encuentra al ser diagnosticado, y si es superficial o profundo. En general, la sobrevida a los 5 años después del diagnóstico suele ser de un 50 %, aunque esta se encuentra estrechamente relacionada con el tipo histológico y estadio del tumor al momento del diagnóstico. Cuando se diagnostica en un estadio inicial y no se ha diseminado a órganos distantes, el tratamiento puede ser muy eficaz y muchos pacientes pueden ser curados; pero, cuando el tumor se ha extendido o ha hecho metástasis en zonas distantes del organismo, el tumor podría incluso ser controlado con el tratamiento, pero no curado. La supervivencia del cáncer debe ser interpretada con mucho cuidado, porque los estimados se basan en miles de casos de este tipo de cáncer, pero el riesgo real de un individuo en particular puede diferir. De ello se deduce que no es posible decir a una persona cuánto va a vivir con el sarcoma.
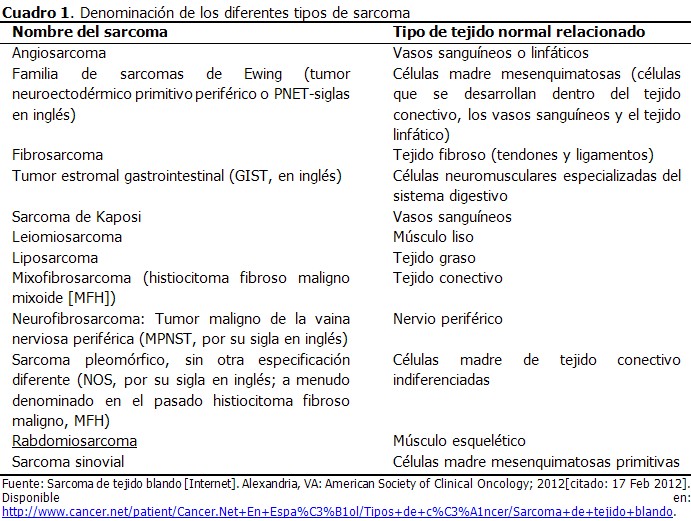
Dado que hay varios tipos diferentes de STS, se trata más bien de una familia de enfermedades relacionadas que de una enfermedad específica única. Los tipos específicos de sarcoma suelen recibir su nombre según las células de tejido normal a las que más se asemejan (Cuadro 1). Esto se diferencia de la mayoría de los demás tipos de cáncer, que generalmente reciben su nombre por la parte del cuerpo en la que el cáncer comenzó.
Los patólogos actualmente intentan identificar nuevos métodos para establecer rápidamente el subtipo de un tumor, ya que esto ayuda a determinar el tratamiento. Examinar la genética anormal de un tumor puede ayudar a determinar sus características y predecir cuáles serán los tratamientos más eficaces.1-3 Quizás, debido a la amplia variedad de sarcomas, casi son inexistentes los textos que incluyan todas las diferentes variedades de este tipo de cáncer, pues generalmente su descripción y revisión se incluyen en las del sistema de órganos afectados específicamente, y por tanto, la literatura que los aborda está muy dispersa en las diferentes especialidades médicas. Por ello, un objetivo primordial en esta revisión es lograr unir la información más actual disponible acerca de la etiología y síntomas de los sarcomas.
Método de localización, selección y evaluación de estudios primarios
Se realizó primeramente una revisión de literatura impresa en libros de patología clínica, oncología clínica y quirúrgica en la biblioteca del Instituto Nacional de Oncología y Radiología (INOR), en la Hemeroteca Nacional y en la biblioteca del Hospital Clínico-Quirúrgico Freyre de Andrade. La revisión de la literatura impresa permitió obtener datos sobre epidemiología, clasificación y anatomía patológica. También fue de gran utilidad para precisar los términos de búsqueda que se emplearon posteriormente en las bases de datos electrónicas.
Se consultó con el Dr. Juan Carlos Pérez Cárdenas, profesor auxiliar de Anatomía Patológica de la Escuela Latinoamericana de Medicina, experto en cáncer y dedicado específicamente a los sarcomas en los últimos 10 años; quien nos orientó sobre el enfoque del tema y acerca de las revistas más significativas que lo abordan.
Fueron revisadas 4 revistas médicas especializadas en cáncer: Revista Cubana de Oncología, British Journal of Cancer, Cancer, Molecular Cancer y 2 revistas médicas que incluyeron especialidades de ortopedia (Revista Cubana de Ortopedia y Traumatología) y cirugía (Revista Cubana de Cirugía). La revisión se hizo en base a lo publicado en los últimos 10 años acerca de los sarcomas.
Se realizó búsqueda en las bases de datos PubMed, Cumed, Secimed, Dynamed, BVS, BMN, LiLaCS, Scielo, ESBCO, Cochrane, LIS; y en los sitios electrónicos Medscape.com y Cancer.net. Se utilizaron como palabras clave: sarcoma, rabdomiosarcoma, liposarcoma, kaposi, Edwing, Gist, leiomiosarcoma, angiosarcoma, genética and sarcoma, etiología and sarcoma, biología molecular and sarcomas.
De la literatura disponible, se trabajó con lo publicado en los últimos 5 años. El objetivo fundamental de la búsqueda fue todo lo relacionado con los conocimientos actuales sobre la etiología y manifestaciones clínicas de los diferentes tipos de sarcomas. No se encontraron estudios que englobaran el tema en conjunto, sino por separado para cada tipo de sarcoma.
Como gestor de base de datos se utilizó el programa EndNote X3 v 13.0
DESARROLLO
Etiología de los sarcomas
La mayoría de los sarcomas no tienen causa conocida, pero, en general, los siguientes factores pueden incrementar el riesgo de sufrir esta enfermedad:
- Radioterapia previa. Las personas que han sido sometidas a radioterapia por cáncer tienen un ligero incremento en el riesgo de padecer sarcoma posteriormente.
- Factores genéticos. Las personas con enfermedades hereditarias como Enfermedad de von Recklinghausen (neurofibromatosis), síndrome de Gardner, síndrome de Werner, esclerosis tuberosa, síndrome de carcinoma de células basales nevoides, síndrome de Li-Fraumeni, o retinoblastoma, tienen un alto riesgo de padecer de sarcoma.
- Sustancias químicas. La exposición laboral al monómero de cloruro de vinilo, usado en la elaboración de algunos tipos de plásticos; o a la dioxina, que puede incrementar el riesgo de sufrir sarcoma. Sin embargo, la mayoría de los sarcomas no se asocian a la exposición de tóxicos ambientales.
Los descritos anteriormente son factores de riesgo que incrementan el riesgo de desarrollar cáncer, pero que no son productores de cáncer directamente. Algunas personas que presentan alguno de estos factores, nunca desarrollan cáncer, y por otra parte, personas sin factores de riesgo conocido, sí desarrollan esta enfermedad.1-3
A continuación profundizaremos en los conocimientos más actuales sobre la etiología de los diferentes tipos de sarcomas.
Angiosarcoma.
Es un tumor maligno proveniente de los vasos sanguíneos o linfáticos, conocido también como hemangiosarcoma. Puede involucrar la piel de cabeza y cuello, tejidos blandos profundos de las extremidades y el tronco, y más raramente órganos profundos como el hígado, glándulas suprarrenales, corazón, bazo, huesos (raro) y mamas (raro). Dentro de los sarcomas, tienen una frecuencia tan baja como menos del 1% del total (los casos reportados en la literatura médica son escasos), y se ve con frecuencia similar en ambos sexos con edad entre los 60 y 71 años.4
No han sido encontradas alteraciones genéticas específicas en este tipo de sarcoma, pero sí cariotipos con desajustes complejos que se caracterizan por numerosas ganancias y pérdidas genéticas. Las alteraciones que se encuentran con más frecuencia son: adición, en los cromosomas 5, 8 y 20; y deleción o pérdida en los cromosomas 7, 22 y Y. El crecimiento tumoral es favorecido por factores proangiogénicos.4
El factor de riesgo más importante es el tratamiento con radiaciones, pues se ha observado la aparición de sarcoma en el período post-radioterapia. Los niños que reciben radioterapia en combinación con quimioterapia citostática por otro tipo de cáncer, constituyen un grupo de alto riesgo pudiendo desarrollar esta enfermedad alrededor de un 20-30% del total.
Otro posible factor de riesgo es el síndrome de Stewart-Treves, que no es más que el miembro superior con linfaedema crónico en paciente sometida a mastectomía por cáncer de mama. En este caso se ha atribuido el riesgo de angiosarcoma a la inflamación crónica y al linfaedema crónico.
También son factores de riesgo el linfaedema crónico de origen genético, por trauma e infección a causa de filarias. Además, las fístulas arterio-venosas disfuncionales en pacientes con insuficiencia renal crónica son otro factor de riesgo.
Se ha asociado hemangiosarcoma hepático a la exposición prolongada al cloruro de vinilo, arsénico (usado en pesticidas) y al thorotrast (suspensión coloidal de dióxido de torio usado en épocas pasadas como método de contraste radiológico endovenoso).
Puede aparecer angiosarcoma como complicación en las siguientes condiciones preexistentes:
- Lesiones vasculares benignas como nevos rojos y linfangioma.
- Tumores benignos y malignos de vainas nerviosas.
- Neurofibromas, leiomiomas, hemangioma de células en huso en el síndrome de Maffucci, retinoblastoma, en el Síndrome de Klippel-Trenaunay, xeroderma pigmentoso y en el tumor maligno de células germinales.
- En lesiones del herpes zóster.
- En el síndrome de Aicardi (malformación genética con ausencia del cuerpo calloso).
- Inmunosupresión.4
Sarcoma de Edwing.
Son tumores musculoesqueléticos muy agresivos que aparecen generalmente en huesos planos y huesos largos, casi siempre en adolescentes. El 85 % de los casos se debe a translocación entre los cromosomas 22 y 11, t(11;22) (q24;q12); y el 10 % a translocación entre los cromosomas 22 y 21. Menos del 1 % se origina por translocaciones entre los cromosomas 22 y 7,17 o 2.
Una proporción importante de los sarcomas se produce por translocaciones cromosómicas o vías metabólicas específicas con eventos moleculares característicos. La vía de señalización del factor de crecimiento similar a la insulina (insulin-like growth factor -IGF), está involucrada en la sarcomagénesis y regula el crecimiento celular, la proliferación, supervivencia y transformación de las células. El receptor 1 para el IGF (IGF-1R), es el regulador positivo clave para el sistema del IGF. El receptor IGF-1R es un receptor heterotetramérico que se encuentra unido a la membrana plasmática y se compone de dos subunidades alfa y dos subunidades beta, unidas entre sí por enlaces disulfuro. Al enlazarse el IGF-I e IGF-II a las subunidades alfa, se activan tirosin kinasas dentro de las subunidades beta lo cual conlleva a la autofosforilación y activación de procesos de degradación. El receptor IGF-1R tiene un rol crucial en el crecimiento de la célula tumoral al mediar la mitogénesis y al mantener el fenotipo transformado, protegiendo las células tumorales de la apoptosis o muerte celular y reduciendo las demandas o necesidades de factor de crecimiento. El empeoramiento in vitro de la función del IGF-1R conlleva a una apoptosis a gran escala de las células tumorales y abolición del crecimiento del tumor primario y sus metástasis. La expresión del IGF-1R y sus ligandos está descontrolada en muchos tipos de tumores y su inhibición conlleva a la disminución del crecimiento tumoral y al incremento de la sensibilidad a diferentes tipos de terapias contra el cáncer.5,6
Estudios serios de investigación demuestran que el IGF-1R parece tener un papel central en la iniciación y mantenimiento de varios subtipos de sarcomas. Este receptor ha sido implicado específicamente, con el crecimiento, metástasis y angiogénesis del sarcoma de Edwing y el rabdomiosarcoma.
Fibrosarcoma: sarcoma del tejido fibroso (tendones y ligamentos) más frecuente en niños neonatos y menores de 5 años de edad. También aumenta su frecuencia entre los 10 y 15 años de edad. Son muy agresivos. En las edades más tempranas se les atribuye un origen genético. Como factores de riesgo, pueden citarse las radiaciones y los citostáticos por tumor maligno.7
Tumor estromal gastrointestinal (GIST por sus siglas en inglés)
Antes conocido como miosarcoma gástrico o leiomiosarcoma intestinal, suele presentarse en pacientes mayores de 50 años; la edad promedio es de 58 años. La célula de origen es probablemente la célula intersticial de Cajal, o células madre comunes precursoras de células intersticiales de Cajal y hacia células fenotípicamente de músculo liso. Las células intersticiales de Cajal se encuentran intercaladas entre los nervios autonómicos, y las células musculares lisas. Su principal función es generar el ritmo autónomo de contracciones involucradas en la digestión y peristaltismo, de modo que son también llamadas células marcapaso del tracto gastrointestinal.8
Factores de riesgo: mutaciones heredadas en los exones 8,11,12,13 y 17.
Posibles factores de riesgo son la neurofibromatosis y la tríada de Carney, que es un síndrome tumoral raro en mujeres jóvenes caracterizado por paraganglioma, condroma pulmonar y tumor del estroma gastrointestinal. También en el síndrome de tumor estromal gastrointestinal.
El protooncogen c-kit, localizado en el brazo largo del cromosoma 4, codifica el receptor transmembrana de tirosina kinasa tipo III. El producto de este gen, la proteína kit, tiene un dominio extracelular que es un receptor y un dominio intracelular ligado a una tirosina kinasa. El ligando del receptor es un factor de crecimiento de células precursoras, una vez ligado produce dimerización del receptor, estimulando la fosforilación del dominio kinasa, lo que genera una cascada de señales desde el citoplasma al núcleo, afectando el comportamiento de la célula en su proliferación, diferenciación, adhesión y apoptosis. La expresión de kit (o CD 117 según la terminología estandarizada de antígenos leucocitarios) es extremadamente importante para algunas líneas celulares normales como células precursoras hematopoyéticas, mastocitos, células germinales (espermatogonias y espermátidas), células de Leydig, melanocitos, algunas células epiteliales (del estrato basal de la piel y epiteliales de mama) y también para las células intersticiales de Cajal, células tumorales de Leucemia Mieloide Crónica y GIST.
La expresión de CD 117 ha emergido como el marcador para discriminar el GIST de otros tumores mesenquimáticos gastrointestinales, así su expresión ha pasado a ser casi sinónimo de GIST. Sin embargo, pese a ser altamente sensible es menos específico, ya que puede encontrarse expresado en células tumorales de Sarcoma de Ewing, angiosarcoma, melanoma, cáncer pulmonar de células pequeñas, carcinoma ovárico, linfoma, leucemia, seminoma y neuroblastoma. La caracterización inmunohistoquímica de los GIST incluye además del CD 117 al CD34 (marca positividad alrededor de los ganglios del plexo mioentérico de Aüerbach en el tracto gastrointestinal). Ambos, CD117 y CD34 son positivos en GIST en valores superiores al 90 %. Las secuencias de ADN para c-kit demuestran una alta frecuencia de mutaciones que determinan la activación de la tirosina kinasa en ausencia de la estimulación por su ligando fisiológico, presentando de este modo una proliferación celular aberrante y resistencia a la apoptosis. La mutación más frecuente identificada está en el exón 11 (dominio yuxtamembrana), otras mutaciones en el gen c-kit incluyen defectos en el exón 8, 13 y 17, pero son mucho menos frecuentes. Existe un grupo entre un 5 y un 15 % de los GIST que no expresan el CD 117. En estos pacientes hay mutaciones del receptor alfa del factor de crecimiento derivado de plaquetas o PDGFRA, otra tirosina kinasa, pero este marcador no logra caracterizar a la mayoría de los GIST. Esta mutación provee de un mecanismo oncogénico alternativo a estos tumores.
Hay una asociación entre GIST y neurofibromatosis tipo 1 (NF1). Los pacientes con NF1 son portadores de un GIST en el 7 % de los casos, sin embargo el mecanismo por el cual esto ocurre es desconocido. Se ha intentado reconocer mutaciones en kit y PDGFRA de pacientes con NF1 sin éxito. Sin embargo, en células tumorales de GIST ha sido demostrada la inactivación somática del gen supresor de tumor NF1 debido a una mutación.8
Sarcoma de Kaposi (SK)
Es una neoplasia vascular multifocal cuya presentación característica es a nivel dérmico. Sin embargo, cada una de las 4 variedades descritas (endémica o africana; epidémica relacionada al SIDA; clásica o mediterránea; iatrogénica o postrasplante) no sólo difieren en el curso y pronóstico, sino también en la frecuencia de su presentación. La variedad endémica puede comprometer frecuentemente órganos internos y ganglios linfáticos; la variedad iatrogénica y epidémica suelen presentar compromiso interno o de mucosas precediendo a las lesiones dérmicas.9,10
Es a partir de la epidemia del SIDA y su asociación con el SK que el mundo científico torna su atención sobre esta malignidad. Sobre todo, cuando en 1994 Chang et al10 identificó al herpes virus asociado al SK, o herpes virus humano tipo 8 (HVH-8) en una lesión de SK epidémico. El sarcoma de Kaposi se ha asociado a herpes virus 8, uno de los gamma-herpesvirus más recientemente identificado, y que puede tener un rol etiológico. El ADN del HVH-8 ha sido detectado en todas las variantes del SK y anticuerpos contra un péptido antigénico del KSHV están presentes en el 67 % de pacientes con VIH (virus de inmunodeficiencia humana) con SK y en el 13 % de los pacientes con VIH sin SK. Más del 90 % del DNA de dicho virus se detecta por PCR en las lesiones del SK y casi en el 100 % de los ganglios sensitivos paravertebrales de los pacientes con SK. La seroconversión a anticuerpos contra el antígeno nuclear relacionado con el KSHV-8 se produce antes de la aparición clínica del SK. Actualmente se considera que la vía sexual es la principal ruta de transmisión horizontal para el HVH-8, con una correlación positiva entre el riesgo de adquirir el virus y el número de parejas sexuales, prácticas sexuales específicas (relaciones anales) y una temprana edad de inicio sexual. Esta vía es característica de zonas con baja endemicidad para el HVH-8 y básicamente en población homosexual. Por el contrario, se cree que en zonas de alto endemismo, la transmisión ocurre principalmente entre madre e hijo y entre hermanos. La transmisión sería a través de la saliva, la cual actuaría como un reservorio para el HVH-8. La infección con el HVH-8 es necesaria pero no suficiente para el desarrollo del SK. La edad, género, raza y país de origen son importantes en el individuo infectado por el HVH-8, ya que pueden representar otros factores en la inducción o desarrollo del SK. La transmisión del HHV-8 mediante trasplantes renales es un factor de riesgo en el SK asociado a trasplante. En series de casos publicadas, se ha sugerido una predisposición autosómica recesiva a la aparición del SK clásico en 3 niños, por demás saludables y no relacionados entre sí, con padres consanguíneos.11-14
Liposarcoma
Es el sarcoma más frecuente, constituyendo cerca del 20 % de los sarcomas en adultos y el 1 % del total de diagnóstico de cáncer. Es raro en niños.
Se desconoce su etiología específica. Proviene de elementos mesenquimales primitivos. Es una hipótesis que el trauma es el evento iniciador de la enfermedad. Se han reportado casos de liposarcoma pleomórfico asociados a exposición previa a radiaciones, cicatrices por quemaduras, neurofibromatosis plexiforme generalizada y schwanoma del nervio radial. Factores de riesgo incluyen además la exposición a ciertos productos químicos, el linfaedema crónico (al igual que en el angiosarcoma) y síndromes genéticos predisponentes como retinoblastoma hereditario, síndrome de Li-Fraumeni y neurofibromatosis tipo 1.
Los cambios citogénicos descritos son: translocación t (12;16) o t (12;22) que conducen a fusión de proteínas que contienen terminales-N relacionadas con los genes 1 y 2 acoplándolas con los terminales-C de los miembros de la familia C/EBP. Se piensa que la fusión de proteínas tiene acción en la génesis tumoral. Los liposarcomas indiferenciados se caracterizan por anillos supernumerarios o cromosomas gigantes derivados de la región 12q donde se ha encontrado adición cromosómica. En los liposarcomas de células redondas y en los mixoides, se encuentran mutaciones en el gen supresor tumoral TP53, en alrededor de un 30 % de los casos, así como sobreexpresión de la proteína MDM2. En el análisis molecular del liposarcoma bien diferenciado se encuentra amplificación de los proto-oncogenes HMGI-C, MDM2, y CDK4 con sobreexpresión de las proteínas asociadas a estos.15
Sarcoma estromal endometrial
Tipo de sarcoma proveniente de tejido mesenquimatoso uterino. Se ha relacionado con el uso del fármaco tamoxifeno. Constituyen menos del 5 % de los tumores uterinos.16,17
Rabdomiosarcoma
Suele ser un tumor embrionario derivado del tejido mesenquimal primitivo. Es de gran malignidad y se ve fundamentalmente en niños menores de 6 años de edad. Aparece en músculo estriado que puede incluir al corazón. La pérdida del brazo corto del cromosoma 11 (11p15.5) caracteriza al rabdomiosarcoma embrionario (eRMS). En el rabdomiosarcoma alveolar (aRMS), se producen translocaciones cromosómicas recíprocas t (2;13)(q35;q14) o t (1;13)(p36;q14), que conlleva a fusión quimérica de genes que incluyen el gen PAX3 (cromosoma 2), o el PAX7 (cromosoma 1) y FKHR (cromosoma 13). El resultado final es una fusión genética PAX3-FKHR y PAX7-FKHR que codifica proteínas que a su vez se ligan a dominios transcripcionales, dando lugar a proteínas primitivas y factores transcriptores de mayor potencia. Estas proteínas inducen la transformación celular, inhiben la diferenciación mitogénica y la apoptosis acentuando la actividad oncogénica.18-20
Condrosarcoma
Pueden ser primarios de etiología no conocida (de novo), y secundarios como complicación de una neoplasia benigna de los cartílagos como encondromas u osteocondromas.
Se han encontrado alteraciones génicas como alteraciones del TP53 relacionadas con progresión de un condrosarcoma de bajo grado de malignidad a uno de alto grado. Algunas aberraciones genéticas incluyen el reordenamiento de 12q13–15 y 9p21. Los genes de exostosina (EXT) están involucrados en el condrosarcoma periférico.21
Sarcoma sinovial
Se origina de vainas tendinosas y bursas articulares, no de las articulaciones, y es un tumor de las células sinoviales. Su etiología es desconocida y es de alta malignidad. Se han encontrado múltiples alteraciones genéticas en genes que regulan vías del desarrollo embrionario y la organogénesis. Es un tumor infrecuente.
Osteosarcoma
Sarcoma de tejido sólido. Es el segundo tumor primario maligno de hueso más frecuente después del mieloma múltiple. Su origen es desconocido pero en estudios en animales se ha implicado un origen viral. Factores de riesgo son la presencia de retinoblastoma (incrementa el riesgo aproximadamente en unas 500 veces), las radiaciones ionizantes y mutación p53 así como en niños con historia familiar del síndrome de Li-Fraumen.22
Síntomas de los sarcomas
Angiosarcoma
La presentación clínica depende de la localización del tumor. Si es una lesión de piel puede presentarse como molestia o dolor local pudiendo aparentar una contusión o equimosis que puede progresar a lesiones ulceradas o nodulares. Si la lesión primaria no es cutánea puede presentarse como una tumoración asintomática. Afecta con más frecuencia a los ancianos y las lesiones suelen tener diámetro entre 3-6 cm.
El angiosarcoma primario del bazo puede presentarse con síntomas como dolor en cuadrante superior izquierdo del abdomen (66 % de los casos), fiebre inespecífica, anorexia y pérdida de peso.
El angiosarcoma óseo tiene 2 patrones de presentación: a) tumor que se aparece con múltiples lesiones en un solo hueso, o múltiples lesiones en huesos adyacentes y/o de todos los huesos de una extremidad b) lesión solitaria que progresa rápidamente metastatizando a pulmones y huesos distantes.
El diagnóstico se basa en los estudios radiológicos que incluyen tomografía axial computarizada (TAC) y resonancia magnética nuclear (RMN) y la biopsia. En huesos profundos la biopsia puede ser guiada por TAC.23,24
Sarcoma de Ewing
Los síntomas pueden variar en dependencia de la localización del cáncer. Puede haber dolor localizado de forma intermitente o continua, de ligera intensidad hasta muy severa y se puede acompañar de parestesias. Hay inflamación o una masa palpable. El dolor regional es la razón más frecuente de consulta de los pacientes. En un estudio de cohorte de 47 pacientes los síntomas se comportaron como sigue: dolor regional en un 72 %, masa palpable 11 %, ambos signos en un 15 % y parestesias regionales en un 2 %.25 Otros estudios se comportan de forma similar. El sarcoma de Edwing pélvico se presentó con dolor (88 %) y/o inflamación (36 %) y limitación de los movimientos (33 %) en otro estudio de cohorte de 33 casos registrados por el registro de tumores óseos escocés.26
En algunos casos la forma de presentación puede ser una fractura patológica o espontánea.27
El dolor suele ser nocturno y se puede acompañar de anorexia, pérdida de peso, malestar general y fiebre.
El examen físico puede demostrar sensibilidad local en la zona dolorosa, masa palpable, movimiento articular muy doloroso y limitación de dichos movimientos. Puede demostrarse la fiebre hasta en un tercio de los casos y marcha dolorosa con impotencia funcional si el tumor es de extremidades inferiores. A menudo los casos se confunden con tendinitis u osteomielitis.
Las metástasis ocurren en un 25 % de los casos y suelen ser más frecuentes en pulmones, huesos y médula ósea. La invasión ganglionar es rara.
Los estudios radiográficos que incluyen TAC y RMN ayudan a realizar el diagnóstico. En las radiografías de estructuras óseas se encuentra localización en diáfisis, tumor relacionado con osteolisis, o espícula calcificada en tejidos blandos y/o despegamiento del periostio (formación del triángulo de Codman). En los exámenes humorales se constatan eritrosedimentación elevada y anemia. Una LDH elevada puede orientar a un mal pronóstico. El diagnóstico positivo es mediante biopsia.
Los estudios de inmunohistoquímica son de importancia para diferenciarlo de otros cánceres. Estudios genéticos moleculares pueden ser de utilidad para evaluar el tumor (hibridación con fluorescencia in situ o FISH y transcripción inversa, reacción en cadena de polimerasa o RT-PCR.5,28
Fibrosarcoma
Se presenta como una masa o tumor de tejido blando firme en consistencia con crecimiento rápido, dolorosa y localizada en cualquiera de las extremidades. Puede asociarse a hipoglucemia pues el tumor secreta una sustancia similar a la insulina. El diagnóstico es por biopsia.7,29
Tumor estromal gastrointestinal (GIST)
Son tumores muy heterogéneos que varían en tamaño, morfología y conducta biológica, son un continuo de neoplasias con potencial maligno incierto, comportándose virtualmente como tumores benignos hasta cánceres muy agresivos, muchas veces metastásicos cuando se realiza el diagnóstico. El GIST puede producirse en cualquier punto del tracto gastrointestinal desde el esófago hasta el ano, sin embargo el estómago (39 a 60 %) y el intestino delgado (30 a 42 %) son los sitios más frecuentes de localización de estos tumores. El colon, recto, apéndice, esófago, mesenterio, retro-peritoneo y otros órganos intraabdominales son ubicaciones inhabituales del GIST. El tipo agresivo del GIST se presenta con compromiso local en cavidad abdominal (en peritoneo, mesenterio, omento) o como compromiso a distancia con metástasis hepáticas. Síntomas inespecíficos incluyen una dispepsia hipoesténica caracterizada por saciedad temprana al ingerir alimentos, sensación de abdomen hinchado, sangramiento gastrointestinal y fatiga por anemia. También puede provocar dolor abdominal, anorexia y pérdida de peso. A veces se recoge antecedente familiar del síndrome de tumor estromal gastrointestinal y estos casos pueden presentar hallazgos al examen físico, de la piel como máculas pigmentadas en región perineal, axilas y la cara o una urticaria pigmentosa. El examen físico de abdomen puede demostrar distensión abdominal, ruidos hidroaéreos exacerbados, dolor a la palpación localmente o difusa. La metástasis extraabdominal es rara. Al igual que ocurre en otros tumores metastásicos, la sintomatología del GIST es inespecífica. Sus manifestaciones clínicas más frecuentes son el descenso de pérdida baja de peso y dolor abdominal mal precisado. Puede implantarse en cicatrices quirúrgicas y en trayectos de agujas por paracentesis. El compromiso ganglionar es muy poco frecuente. Son lesiones que frecuentemente se diagnostican por TAC o una endoscopía sugerente. La asociación de endoscopía y ecografía puede ayudar al diagnóstico de certeza, así los GIST son infrecuentes en esófago, por lo que una lesión esofágica subepitelial hipoecogénica de la segunda capa (muscular de la mucosa), o de la cuarta (capa muscular), es más probable que se deba a un leiomioma. En cambio, la misma lesión en el estómago como primera posibilidad corresponde a un GIST. Los estudios con TAC, tomografía con emisión de positrones (PET), RMN, y exámenes endoscópicos, ayudan al diagnóstico clínico. La combinación de la ecografía abdominal con PAAF (punción por aspiración con aguja fina) puede permitir obtener una muestra adecuada para realizar un diagnóstico de GIST.8
Sarcoma de Kaposi
Se presenta como nódulos (umbilicados o no), máculas o pápulas purpúricas que pueden aparecer clásicamente en la piel de las extremidades inferiores, pero puede involucrar órganos internos y ganglios linfáticos comprometiendo con menor frecuencia los músculos, sistema nervioso central, nervios periféricos, corazón, mamas y los ojos. Cerca del 60 % de los casos tienen lesiones en piel y mucosa oral. El otro 40 % tiene compromiso del tracto gastrointestinal o invasión pulmonar, y pueden cursar con sangramiento digestivo crónico o agudo, perforación u obstrucción. El estudio endoscópico de estos casos muestra las características lesiones nodulares purpúricas o lesiones polipoides. Si el paciente es portador de VIH/SIDA puede cursar con los síntomas y signos que conforman todo el espectro de dicha enfermedad, siendo el tumor maligno más frecuente que se asocia al SIDA. En una serie de casos de pacientes con SIDA de Lebwohl y cols. se encontró compromiso visceral digestivo en el 50 % de los casos, invasión de ganglios linfáticos en el 50 % y toma pulmonar en el 37 %. La presencia de Sarcoma de Kaposi en paciente con VIH positivo, lo define como enfermo de SIDA. El diagnóstico se realiza mediante biopsia de las lesiones.29-36
Liposarcoma
Pueden aparecer en cualquier lugar del organismo. Existen 4 subtipos histológicos: pleomórfico, bien diferenciado, mixoide e indiferenciado. Muslos y retroperitoneo son los lugares de mayor frecuencia. Constituye uno de los sarcomas más frecuentes en las extremidades. Otros sitios menos frecuentes son lengua, estómago, esófago, hueso, vesícula biliar, parótidas, pies, mamas, pleura, tiroides, espacio retrofaríngeo, hígado, paratesticular y cavidad oral.
Se manifiestan como masa indolora, y con síntomas en dependencia de la localización, relacionados con presión local aumentada (efecto de masa), edema distal a la lesión, síntomas vesicales y parestesias.
El liposarcoma retroperitoneal puede cursar con dolor, tumor abdominal, molestias gastrointestinales inespecíficas, distensión y pérdida de peso. Síntomas infrecuentes son el sangramiento gastrointestinal, obstrucción intestinal parcial y síntomas neurológicos relacionados con invasión o compresión de estructuras neurovasculares. Puede que se palpe tumor firme e indoloro, que puede incluso ser muy bien delimitado a la palpación. Puede adquirir grandes dimensiones en el espacio retroperitoneal. El diagnóstico es por biopsia del tumor.37-39
Sarcoma estromal endometrial
Puede indagarse en el interrogatorio acerca del uso de tamoxifeno en algún momento de los últimos 5 años. Suele presentarse más frecuentemente con sangramiento vaginal irregular y/o como una masa tumoral pélvica.
El examen físico solo aporta un tumor uterino discreto o un agrandamiento uterino difuso. El examen físico y estudios complementarios definirán el estadio al diagnóstico.
- Estadio I - enfermedad confinada al cuerpo uterino.
- Estadio II - enfermedad que involucra el cérvix.
- Estadio III - extensión a órganos pélvicos.
- Estadio IV - enfermedad con extensión fuera de la pelvis.16,17,40,41
Rabdomiosarcoma
Se manifiesta como tumor o masa palpable, ocasionalmente como masas voluminosas redondeadas polipoides que pueden proyectarse hacia afuera como una invaginación. Puede tener forma nodular. Los estudios radiológicos y la biopsia definirán el diagnóstico, al igual que en otras variedades de sarcoma.19,41
Condrosarcoma
Clínicamente, se presenta como dolor profundo en el sitio del tumor y/o masa palpable. Suelen ser de crecimiento lento y rara vez metastatizan teniendo un excelente pronóstico después de una cirugía adecuada. El diagnóstico se basa en estudios radiológicos y la biopsia. El tipo histológico influye en el pronóstico, pues la variedad indiferenciada y la mesenquimal son altamente agresivas. En la variedad de células claras pueden ocurrir metástasis hasta 24 años después del diagnóstico inicial y su tratamiento.43
Sarcoma sinovial
Los sitios involucrados suelen ser extremidades inferiores (66 %) y superiores (34 %). Se ha reportado un caso que debutó con tumor palpable del cuello. Es más frecuente en hombres de cualquier edad pero con un pico de incidencia entre los 30-50 años de edad. Los síntomas y signos son locales, con dolor articular, y el tumor suele ser profundo. Son alto grado de malignidad pudiendo metastatizar en pulmón y pleura y se producen recurrencias locales frecuentemente. El diagnóstico positivo se realiza mediante biopsia.42
Osteosarcoma
Los síntomas suelen ser dolor óseo, inflamación local y fracturas patológicas. Síntomas generales como fiebre, anorexia, palidez y pérdida de peso, son infrecuentes.
Las metástasis afectan con frecuencia a pulmón, hígado y cerebro.
Los estudios radiológicos pueden mostrar la fractura ósea del hueso estudiado, y el triángulo de Codman, que puede extenderse hasta el tejido blando aledaño a la
lesión. La fosfatasa alcalina del suero puede elevarse en cifras de 2-3 veces su valor normal. Un factor de crecimiento endotelial vascular, presente en más del 25 % de las células tumorales obtenidas por biopsia, se relaciona con menor supervivencia del paciente.43-46
CONCLUSIONES
Los sarcomas son un grupo de tumores malignos que se originan en los tejidos que dan soporte y conexión al cuerpo (cáncer del tejido mesenquimatoso). Pueden aparecer en cualquier parte del cuerpo. Las células del sarcoma se parecen a aquellas que mantienen unido el cuerpo, incluidas las células grasas, los músculos, los nervios, los tendones, las articulaciones, los vasos sanguíneos o los linfáticos.
En general son tumores de gran agresividad y de sintomatología muy variada en dependencia del tipo de sarcoma y estadio tumoral al momento del diagnóstico.
Además de los síntomas generales, suelen cursar todos con dos signos primordiales: dolor y aumento de volumen.
Aunque en muchos no se conoce con certeza la etiología, se han identificado diferentes factores de riesgo y factores etiológicos, tanto genéticos, infecciosos, como ambientales, sobre los que se puede actuar tanto con el objetivo de lograr una prevención primaria (evitar la aparición del tumor aboliendo la exposición a productos tóxicos como el cloruro de vinilo, el thorotrast etc. o radiaciones ionizantes, campaña de prevención del VIH/SIDA), como una prevención secundaria( tratar de realizar diagnósticos precoces en pacientes de alto riesgo de desarrollar sarcoma como pacientes previamente sometidos a radioterapia o con síndromes genéticos predisponentes o portadores de VIH/SIDA).
Si bien queda mucho por avanzar en el conocimiento de la etiología de los sarcomas, los grandes descubrimientos en relación con los mecanismos genéticos involucrados en los diferentes tipos de sarcoma han abierto un camino de inestimable valor para introducir nuevos tratamientos que incluyen ensayos con anticuerpos monoclonales y nuevos fármacos de terapia génica. El estudio genético ha permitido también introducir muchas herramientas a nivel molecular, de gran utilidad en el diagnóstico y pronóstico del tipo de sarcoma en cuestión. Por esta razón, es de gran importancia profundizar en el estudio de la etiología de los sarcomas, a lo cual contribuye el presente estudio, aportando una compilación de los conocimientos más actuales sobre este aspecto acerca de los sarcomas.
REFERENCIAS BIBLIOGRÁFICAS
- Kumar V, Abbas AK, Fausto N, Aster J. Robbins and Cotran Pathologic Basis of Disease. 8th ed. Philadelphia: W. B. Saunders; 2006 [Buscar en Google Scholar]
- McGuire WW. Clinical Evidence Concise. London: BMJ Publishing Group; 2004 [Buscar en Google Scholar]
- Gadgeel SM, Harlan LC, Zeruto CA, Osswald M, Schwartz AG. Patterns of care in a population based sample of soft tissue sarcoma patients in the United States. Cancer. 2009;115(12):2744-54 [Buscar en Google Scholar]
- Angiosarcoma [Internet]. Ipswich (MA) : EBSCO Publishing; 2012 [citado 7 Mar 2012]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=aaaf4233-b2e0-486d-b6ce-13cccdaedfc9@sessionmgr15&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=116282 [Buscar en Google Scholar]
- Kohnle D, Kellicker PG. Sarcoma de Ewing: Tumores neuroectodérmicos primitivos [TNEP] periféricos; familia de tumores de Ewing [Internet]. Nashville: MEDtropolis; 2010 [citado Mar 12]. Disponible en: http://healthlibrary.epnet.com/GetContent.aspx?token=b696f142-edf4-4d7f-a7ba-dc89ceee0ace&chunkiid=247778&siteid=82cb1fab-8e7e-11d4-81f3-00508b1249d5 [Buscar en Google Scholar]
- Arora S, Gonzales IM, Hagelstrom RT, Beaudry C, Choudhary A, Sima C ,et al. RNAi phenotype profiling of kinases identifies potential therapeutic targets in Ewing s sarcoma. Mol Cancer. 2010;9:218 [Buscar en Google Scholar]
- Fibrosarcoma [Internet]. Ipswich (MA): EBSCO Publishing; 2011 [citado 7 Mar 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=366c6ab2-fdce-496f-8ec7-9a90211ce110@sessionmgr12&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=116515 [Buscar en Google Scholar]
- Pablo Bórquez M, Rodrigo Neveu C. Tumores del estroma gastrointestinal (GIST), un particular tipo de neoplasia. Rev Méd Chile [revista en Internet]. 2008 [citado 12 Mar 2011];136(7):[aprox. 16p]. Disponible en: http://www.scielo.cl/scielo.php?pid=S0034-98872008000700016&script=sci_arttext [Buscar en Google Scholar]
- Kaposi sarcoma [Internet]. Ipswich (MA): EBSCO Publishing; 2012 [citado 7 Mar 2012]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=980d189a-15f2-4025-91fd-1f4feabd1fd4@sessionmgr14&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU= [Buscar en Google Scholar]
- Alan R. Sarcoma de Kaposi [Internet]. Boston: Tufts Medical Center; 2011 [citado 12 Mar 2011]. Disponible en: http://www.tuftsmedicalcenter.org/apps/HealthGate/Article.aspx?chunkiid=103617 [Buscar en Google Scholar]
- Dittmer DP, Krown SE. Targeted therapy for Kaposi’s sarcoma and Kaposi s sarcoma associated herpesvirus. Curr Opin Oncol. 2007;19(5):452-7 [Buscar en Google Scholar]
- Rezza G, Andreoni M, Dorrucci M, Pezzotti P, Monini P, Zerboni R ,et al. Human herpesvirus 8 seropositivity and risk of Kaposis sarcoma and other acquired immunodeficiency síndrome - related disease. J Natl Cancer Inst. 1999;91(17):1468-74 [Buscar en Google Scholar]
- Lee HR, Lee S, Chaudhary PM, Gill P, Jung JU. Immune Evasion by Kaposi s Sarcoma-associated Herpesvirus. Future Microbiol. 2010;5(9):1349-65 [Buscar en Google Scholar]
- Wang L, Dittmer DP, Tomlinson CC, Fakhari FD, Damania B. Immortalization of primary endothelial cells by the K1protein of Kaposi s sarcoma-associated herpesvirus. Cancer Res. 2006;66(7):3658-66 [Buscar en Google Scholar]
- Liposarcoma [Internet]. Ipswich (MA): EBSCO Publishing; 2012 [citado 7 Mar 2012]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=5ea46b5f-bc94-43d8-a1b7-8bfebfaae1c3@sessionmgr14&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU= [Buscar en Google Scholar]
- Endometrial stromal sarcoma [Internet]. Ipswich (MA): EBSCO Publishing; 2011 [citado 7 Mar 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=bb0af78b-646f-4cb8-b942-f96ee1322c09@sessionmgr4&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=116296 [Buscar en Google Scholar]
- Uterine leiomyosarcoma [Internet]. Ipswich (MA) : EBSCO Publishing; 2011 [citado 28 Nov 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=8b45e54e-3f2f-435a-845f-1aeb7015f543@sessionmgr4&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU= [Buscar en Google Scholar]
- Alveolar rhabdomyosarcoma [Internet]. Ipswich (MA): EBSCO Publishing; 2011 [citado 28 Nov 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=c0b1ede5-ed52-4272-88e9-54d143e9b703@sessionmgr4&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU= [Buscar en Google Scholar]
- Embryonal rhabdomyosarcoma [Internet]. Ipswich (MA) : EBSCO Publishing; 2011 [citado 28 Nov 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=5b20d309-0519-4b7b-807a-39d6094b2148@sessionmgr4&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU= [Buscar en Google Scholar]
- Hu K, Lee C, Qiu D, Fotovati A, Davies A, Abu-Ali S, et al. Small interfering RNA library screen of human kinases and phosphatases identifies polo-like kinase 1 as a promising new target for the treatment of pediatric rhabdomyosarcomas. Mol Cancer Ther. 2009;8(11):3024-35 [Buscar en Google Scholar]
- Synovial Sarcoma [Internet]. Ipswich (MA) : EBSCO Publishing; 2012 [citado 2 Abr 2012]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=19865b4c-82b2-4839-bbfd-8c7afb41a95f@sessionmgr11&vid=1&hid=15&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=115341 [Buscar en Google Scholar]
- Osteogenic sarcoma [Internet]. Ipswich (MA) : EBSCO Publishing; 2011 [citado 28 Feb 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=1db85713-0de1-4c55-88d3-4d6de6d22ce7@sessionmgr111&vid=1&hid=113&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=116168&anchor=anc-1965744510 [Buscar en Google Scholar]
- Radulescu D, Pripon S, Radulescu LI, Constantea NA, Gulei I. A rare case of primitive right atrium angio-sarcoma with favorable outcome, in a young female: Case report and literature review. Rev Med Chil. 2008;136(10):1311-6 [Buscar en Google Scholar]
- Cordies Justin NE, Cordies Justin R. Sarcomas de partes blandas. Estudio comparativo de dos decenios. Rev Cubana Oncol. 1997;13(1):31-6 [Buscar en Google Scholar]
- Widhe B, Widhe T. Initial Symptoms and Clinical Features in Osteosarcoma and Ewing Sarcoma. J Bone Joint Surg Am. 2000;82(5):667-74 [Buscar en Google Scholar]
- Bhagat S, Sharma H, Pillai DS, Jane MJ. Pelvic Ewing s sarcoma: a review from Scottish Bone Tumour Registry. J Orthop Surg (Hong Kong). 2008;16(3):333-8 [Buscar en Google Scholar]
- Jackson WF, Theologis TN, Gibbons CL, Mathews S, Kambouroglou G. Early management of pathological fractures in children. Injury. 2007;38(2):194-200 [Buscar en Google Scholar]
- Codman EA. The classic: registry of bone sarcoma: part I.--Twenty-five criteria for establishing the diagnosis of osteogenic sarcoma. Part II.--Thirteen registered cases of five year cures analyzed according to these criteria. 1926. Clin Orthop Relat Res. 2009;467(11):2771-82 [Buscar en Google Scholar]
- Francis P, Namlos HM, Muller C, Edén P, Fernebro J, Berner JM, et al. Diagnostic and prognostic gene expression signatures in 177 soft tissue sarcomas: hypoxia-induced transcription profile signifies metastatic potential. BMC Genomics. 2007;8:73 [Buscar en Google Scholar]
- Wang L, Korman A, Favila K. Kaposi s Sarcoma of the Upper GI Tract Causing Severe Anemia. Am J Gastroenterol. 2011;106(2S):S372 [Buscar en Google Scholar]
- Albín Cano RG, Perez Cardenas JC. Conjunctival metastasis from Kaposi s sarcoma: a case report. Diagn Cytopathol. 2011;39(2):128-31 [Buscar en Google Scholar]
- Newton R, Carpenter L, Casabonne D, Beral V, Babiker A, Darbyshire J, et al. A prospective study of Kaposi s sarcoma-associated herpesvirusand Epstein–Barr virus in adults with human immunodeficiency virus-1. Br J Cancer. 2006;94(10):1504-9 [Buscar en Google Scholar]
- Iscoe N, Bramwell V, Charette M, Oliver T, Zanke B; Systemic Treatment Disease Site Group. Liposomal anthracyclines in the management of patients with HIV-positive Kaposi s sarcoma [Internet]. Reino Unido: Universidad de York; 2006 [citado 7 Mar 2011]. Disponible en: http://www.crd.york.ac.uk/CRDWeb/ShowRecord.asp?ID=12003008549 [Buscar en Google Scholar]
- Pantanowitz L, Dezube BJ. Kaposi sarcoma in unusual locations. BMC Cancer. 2008;8:190 [Buscar en Google Scholar]
- Dedicoat M, Vaithilingum M, Newton R. Treatment of Kaposis sarcoma in HIV-1 infected individuals with emphasis on resource poor settings. Cochrane Database Syst Rev. 2003;(3):CD003256 [Buscar en Google Scholar]
- Roiz Balaguer M, Morales Barrabia I. Sarcoma de Kaposi: clasificación y evaluación en Zimbabwe . Rev haban cienc méd. 2010 [citado 12 Abr 2011];9(2):[aprox. 13p]. Disponible en: http://scielo.sld.cu/scielo.php?pid=S1729-519X2010000200013&script=sci_arttext. [Buscar en Google Scholar]
- Canter RJ, Qin LX, Ferrone CR, Maki RG, Singer S, Brennan MF. Why Do Patients with Low Grade Soft Tissue Sarcoma Die?. Ann Surg Oncol. 2008;15(12):3550-60 [Buscar en Google Scholar]
- Vázquez González JM, Danta Fundora D, Collado Otero JC, Paredes López D. Liposarcoma mediastínico metastásico. Rev cuba Cir [revista en Internet]. 2009 [citado 12 Abr 2011];48(3):[aprox. 12p]. Disponible en: http://www.bvs.sld.cu/revistas/cir/vol48_3_09/cir08309.pdf [Buscar en Google Scholar]
- Badash M. Sarcoma de tejido blando [Internet]. Lowell: EBSCO Publishing; 2011 [citado 12 Dic 2011]. Disponible en: http://healthlibrary.epnet.com/GetContent.aspx?token=981f9709-f625-4a42-a685-d1cde949efa5&chunkiid=103479 [Buscar en Google Scholar]
- Avila Arostegui DL, Amores Carraté J, Bastián Manso L, Arredondo Bruce A. Tumor maligno mülleriano mixto: a propósito de un caso raro. AMC [revista en Internet]. 2010 [citado 12 Abr 2011];14(5):[aprox. 10p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1025-02552010000500014 [Buscar en Google Scholar]
- Malignant mixed mullerian tumor of endometrium [Internet]. Ipswich (MA) : EBSCO Publishing; 2011 [citado 12 Abr 2011]. Disponible en: http://web.ebscohost.com/dynamed/detail?sid=9038214e-995d-47f0-a594-92b2cffcb2d3@sessionmgr115&vid=3&hid=113&bdata=JnNpdGU9ZHluYW1lZC1saXZlJnNjb3BlPXNpdGU=#db=dme&AN=115789 [Buscar en Google Scholar]
- Zamani F, Jabbari M, Alimohamadi SM, Shakeri R, Rostami Z, Abedi B, et al. Primary alveolar soft part sarcoma of chest wall: a case report and review of the literature. Med Gen Med. 2006;8(3):2 [Buscar en Google Scholar]
- McCoy K. Cáncer de los huesos [Internet]. Virginia: EBSCO Publishing; 2010 [citado 12 Dic 2011]. Disponible en: http://www.aurorahealthcare.org/yourhealth/healthgate/getcontent.asp?URLhealthgate="103792.html" [Buscar en Google Scholar]
- Valdivia Ninel R, Gómez Goliath R, Cárdenas Centeno OM, Sánchez Noda EO. Sarcoma granulocítico del tercio distal del fémur. Presentación de 1 caso inusual. Rev Cubana Ortop Traumatol. 1999;13(1-2):123-28 [Buscar en Google Scholar]
- Álvarez López A, García Lorenzo Y, Puentes Álvarez A, García Lorenzo M. Osteosarcoma: enfoque actual. AMC [revista en Internet]. 2010 [citado 19 May 2011];14(5):[aprox. 13p]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S1025-02552010000500016 [Buscar en Google Scholar]
- Picci P. Osteosarcoma (osteogenic sarcoma). Orphanet J Rare Dis. 2007;2:6 [Buscar en Google Scholar]
Enlaces refback
- No hay ningún enlace refback.

FINLAY EN: 








FINLAY CERTIFICADA POR:

Esta revista "no aplica" cargos por publicación en ninguna etapa del proceso editorial.
Dirección postal: Calle 51A y Avenida 5 de Septiembre Cienfuegos, Cuba Código postal: 55100.
http://www.revfinlay.sld.cu
Telefono: +53 43 516602. Telefax: +53 43 517733.
amgiraldoni@infomed.sld.cu
ISSN: 2221-2434
RNPS: 5129